Properties of liquids interacting with pressurized gases: from natural gas to hydrogen economy
Call: 34th Open Access Grant Competition; OPEN-34-13
Researcher: Jan Heyda
Institution: University of Chemistry and Technology Prague
Field: Material sciences

The dissolution of gaseous methane (under high pressure of 105 bar) and its diffusion in liquid p-xylene take place inside a 9 mm titanium measuring cell. The temporal evolution of these processes, including the position and shape of the interface (green and pink curves), is monitored using a unique neutron imaging method. Molecular dynamics simulations provide a sub-nanometre view of the arrangement of methane in the gas phase (black spheres), at the interface (blue), and within (yellow) the liquid p-xylene (turquoise region). By combining experiments and simulations, it becomes possible to evaluate key physico-chemical properties of these systems under conditions closely resembling those encountered in the energy industry.
Scientists from the University of Chemistry and Technology in Prague will harness the power of the Barbora and Karolina supercomputers to investigate how selected liquids interact with gases such as methane, ethane and hydrogen under high pressure. These insights are crucial both for today’s natural gas industry and for the emerging hydrogen economy. In current energy systems, methanol serves as an inhibitor of undesirable methane hydrate formation.
Looking ahead, methanol and toluene are considered promising liquid organic hydrogen carriers (LOHCs) for future energy infrastructures. Using molecular simulations, the research team is examining non-bonded interactions in detail, thereby complementing unique neutron imaging of high-pressure systems carried out in collaboration with scientists from the Paul Scherrer Institute (PSI).
The research forms part of the Czech–Swiss project “Properties of Liquids Exposed to Compressed Methane, Ethane and Hydrogen” (GA ČR–SNSF 23-04741K).
Exploring the mechanisms of plasticity in nitinol
Call: 34th Open Access Grant Competition, OPEN-34-88
Researcher: Miroslav Černý
Institution: CEITEC
Field: Material Sciences
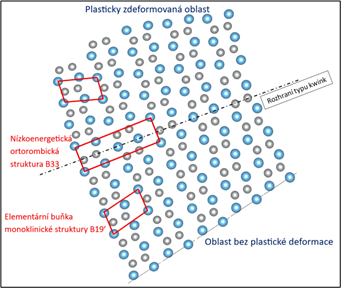
An atomistic simulation of plastic deformation in the martensitic structure of Nitinol. Within this structure, researchers have identified a new plastic deformation mechanism – known as kwinking (a term coined by combining twinning and kinking) – which merges the processes of twinning and anisotropic plastic slip.
A research team from CEITEC, Brno University of Technology and the Czech Academy of Sciences is harnessing the power of the Karolina, LUMI and Barbora supercomputers to study in detail the properties of a nickel–titanium alloy – Nitinol (NiTi). Thanks to its ability to return to its original shape after deformation and subsequent heating – a phenomenon known as shape memory – Nitinol is a unique material with a wide range of applications, from medical devices to technical components. In addition to its shape memory effect, Nitinol also displays a surprisingly high ductility, i.e. the capacity for plastic deformation.
This remarkable combination of properties stems from mechanisms at the atomic level that remain largely unexplored. To uncover them, scientists are employing novel approaches in atomic simulations based on machine learning and quantum-mechanical calculations, which now make it possible to reconstruct the behaviour of materials down to the level of individual atoms.
The research, carried out within the Czech Science Foundation project GA ČR 25-16285S, “Kwinking concept in NiTi shape memory alloy”, paves the way for the development of new materials that combine the unique features of shape memory alloys with the high toughness of materials utilising TRIP and TWIP mechanisms.
Machine learning and semi-empirical quantum chemistry
Call: 33rd Open Access Grant Competition; OPEN-33-68
Researcher: Jan Řezáč
Institution: Institute of Organic Chemistry and Biochemistry of the Czech Academy of Sciences
Field: Material Sciences
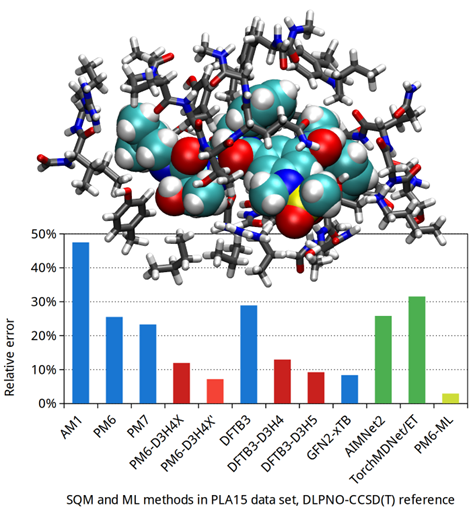
Jan Řezáč from the Czech Academy of Sciences of the Czech Republic will use the Karolina and Barbora supercomputers to develop a new method in computational chemistry that combines semiempirical quantum mechanical (SQM) computation with machine learning (ML). The result will be a tool that offers the accuracy of density functional theory (DFT)-based methods but with significantly lower computational requirements. The method's current version, PM6-ML, has already been published in the Journal of Chemical Theory and Computation.
With the support of supercomputers, the research team is now working on improving the machine learning model and expanding the training dataset. The goal is to create an even more accurate and versatile method that retains excellent scalability for large-scale computations, especially for applications in drug development. Based on open-source software, the tool under development is already freely available to the scientific community. The research is supported by the Grant Agency of the Czech Republic.
Strategies of chemical doping for high-efficiency thermoelectricity in transition metal nitrides
Call: 32nd Open Access Grant Competition, OPEN-32-7
Primary Investigator: Luigi Cigarini and Dominik Legut
Institution: IT4Innovations
Research Area: Material Sciences
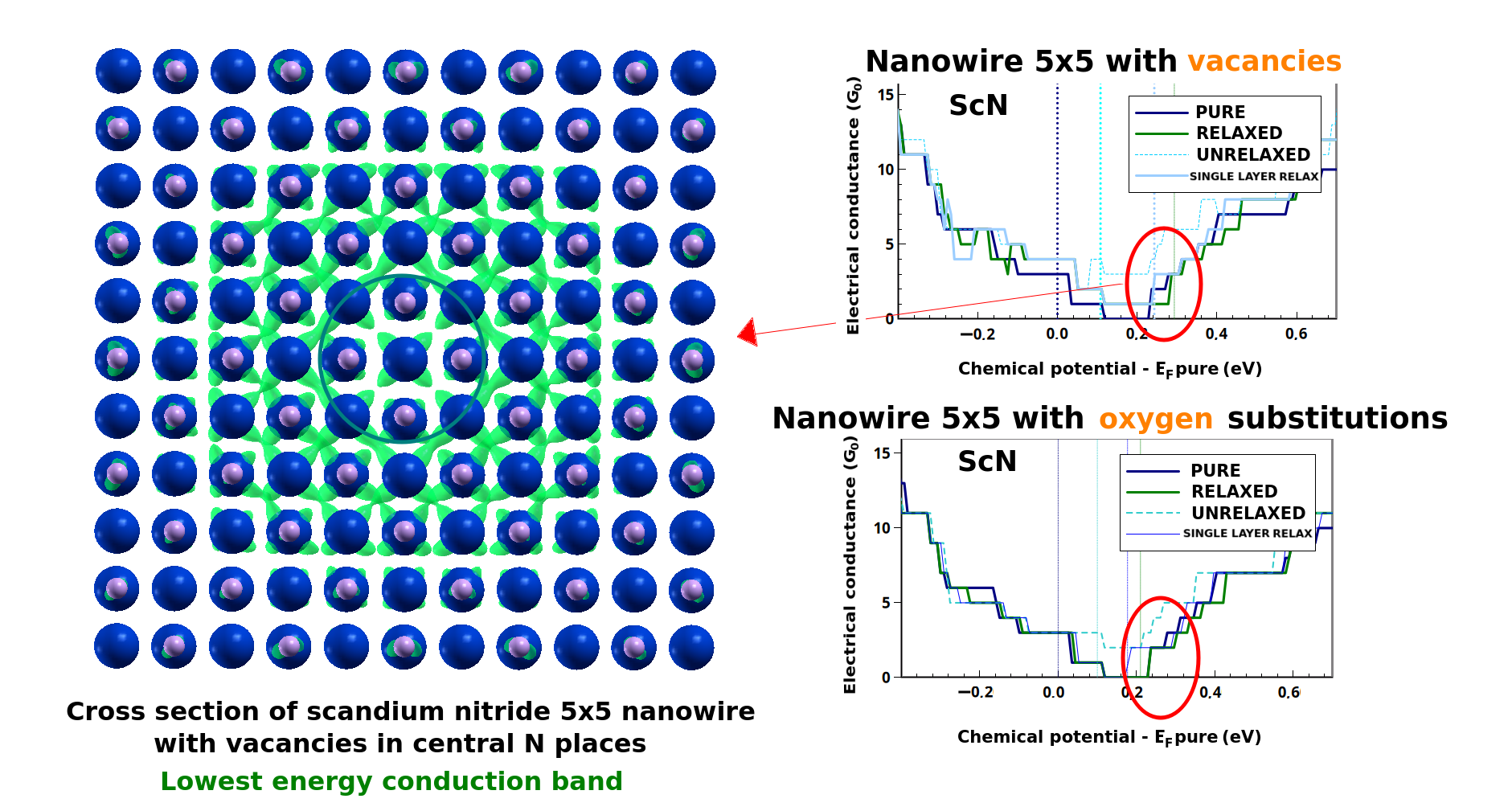
The image shows a comparison of the effects on electron transport caused by atomic vacancies and substitutional oxygen atoms replacing the central nitrogen atoms in an infinite nanowire structure composed of 5x5 crystal cells of scandium nitride.
Luigi Cigarini, Urszula D. Wdowik, and Dominik Legut from IT4Innovations will utilize the computational power of the Barbora, Karolina, and LUMI supercomputers to explore the thermoelectric properties of scandium nitride and chromium nitride — two promising materials for energy conversion technologies. The theoretical calculations will focus on the effects of defects and dopants in the crystal structures of these materials.
Thermoelectricity, the ability to convert temperature differences into electric voltage and vice versa, holds significant potential for improving energy efficiency in industries, vehicles, and household devices. The project aims to develop strategies to enhance the performance of cost-effective thermoelectric materials, paving the way for innovative energy solutions and advancing technological progress in energy efficiency. The research is supported by the Czech Science Foundation by the grant No. 23-07228S.
Covalent Dative Bonding, H-Bonding, and Charge Transfer Complexes: Surprising Stability/Instability Trends with Increasing Solvent Polarity
Call: 31st Open Access Grant Competition; OPEN-31-51
Researcher: Pavel Hobza
Institution: Institute of Organic Chemistry and Biochemistry
of the Czech Academy of Sciences and IT4Innovations
Field: Material sciences
The project of Professor Pavel Hobza from the Institute of Organic Chemistry and Biochemistry of the CAS and IT4Innovations focuses on studying the effect of solvent polarity on the stability of covalent dative and non-covalent complexes.
To analyse these interactions, Professor Hobza's team will use the Karolina, Barbora, and LUMI supercomputers and modern laboratory methods. Solvation, the envelopment of ions by solvent molecules, plays a crucial role in supramolecular chemistry.
The research aims to gain a deeper understanding of the chemical bonds and interactions in different solvent environments, leading to the development of new technologies and materials necessary for solvation processes.
Many-body physics of van der Waals heterostructures from 2D materials
Call: 31st Open Access Grant Competition; OPEN-31-8
Researcher: František Karlický
Institution: University of Ostrava
Field: Material sciences
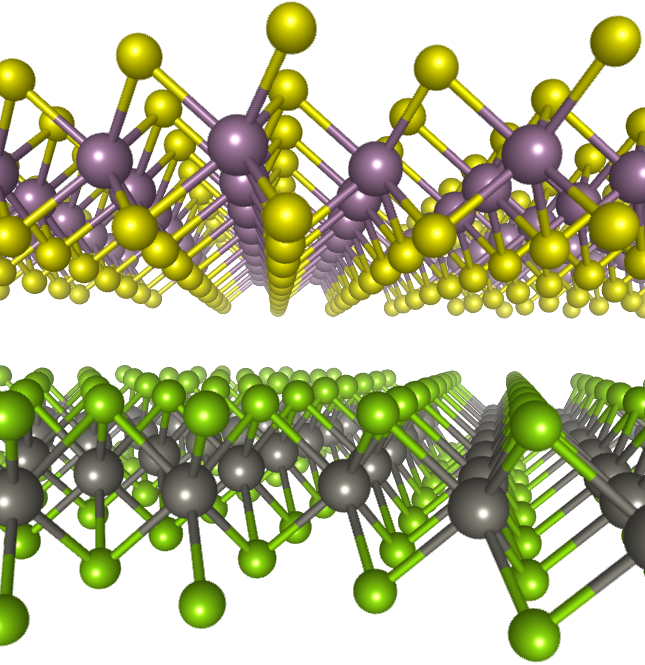
František Karlický and his team of nanostructure physicists from the University of Ostrava (nano.osu.cz) focus, among other things, on two-dimensional (2D) materials that have the potential for flexible and ultrathin functional devices such as future nanoelectronics and solar cells. They also investigate van der Waals (vdW) heterostructures formed by layering 2D materials and have unique electronic and optical properties.
They use computationally intensive multiparticle methods to simulate physical phenomena that traditional methods cannot capture. This research, funded by the Just Transition Operational Programme (LERCO), will contribute to a better understanding of vdW heterostructures and support further experimental research and technological applications.
Optimizing ARPES experiments through AI: Achieving precise photon beam polarization with graphene calibration
Call: 31st Open Access Grant Competition; OPEN-31-35
Researcher: Ridha Eddhib and Ján Minár
Institution: University of West Bohemia
Field: Material sciences
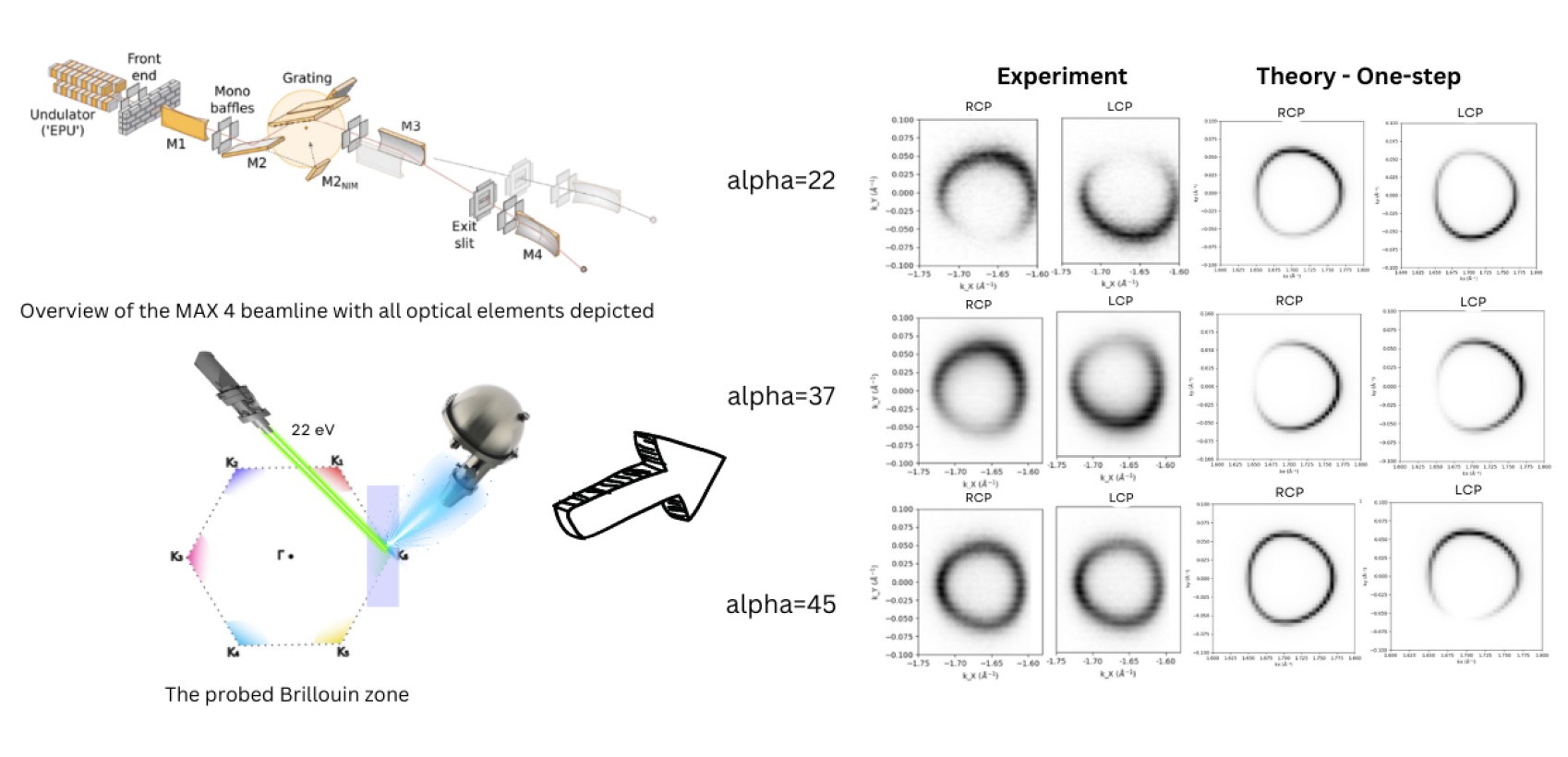
Graphene and polarization detection: The figure shows the MAX 4 beamline setup, illustrating the key optical components involved. The probed Brillouin zone is highlighted, with a photon energy of 22 eV. The right side presents experimental circular dichroism patterns for different angles (alpha = 22, 37, and 45 degrees) using right- and left-circularly polarized light (RCP and LCP). These experimental results are compared with theoretical predictions, demonstrating good agreement across the different angles.
The EuSpecLab-ML supercomputing project, led by Prof. Ján Minár and his PhD student Ridha Eddhib from the New Technologies Research Center (NTC), University of West Bohemia, is set to revolutionize Angle-Resolved Photoemission Spectroscopy (ARPES) by harnessing the power of Machine Learning (ML) techniques. ARPES is a critical technique in condensed matter physics, providing deep insights into the electronic structures and quantum states of materials.
However, the precision of ARPES measurements critically depends on the accurate calibration of the photon’s beam polarization – a challenge that has long confronted experimentalists.
In this project, researchers aim to achieve 100% polarization accuracy by developing an advanced Neural Network (NN) model, based on PyTorch library. This model will be meticulously trained on datasets generated by the sophisticated one-step model of the SPRKKR (Spin-Polarized Relativistic Korringa-Kohn-Rostoker) code, renowned for its accurate ARPES simulation capabilities. The ase2sprkkr package will be employed for seamless interfacing with SPRKKR, enriching the AI model’s training data with high-fidelity simulations of graphene – a 2D material celebrated for its exceptional electronic properties and pivotal role in material science. To streamline and enhance the efficiency of the model training process, the project introduces SparkkFLOW, a novel workflow tailored for managing and analyzing ARPES simulation data. Coupled with the JobMaster workflow manager, SparkkFLOW enables the automated generation, organization, and sorting of simulation data, significantly reducing the time and computational resources required for model training. This integration of AI with cutting-edge computational tools allows for real-time prediction of optimal ARPES experimental conditions, ensuring that each measurement is conducted under the most precise polarization conditions.
The research leverages the high-performance computing resources of the supercomputer Karolina, part of the European High-Performance Computing Joint Undertaking (EuroHPC JU). The ultimate goal is to provide experimentalists with a robust tool to dynamically adjust their ARPES setups, thereby optimizing beamline time and enhancing efficiency at synchrotron facilities. Funded by the MSCA Horizon project and OPJAK, forming part of the EuSpecLab Doctoral Training Network, this research not only aims to elevate the quality and accuracy of ARPES experiments but also opens new avenues for exploring the electronic behaviors of advanced materials, including novel 2D systems and quantum materials. project is funded by OPJAK: Researchers are thankful for the support of the QM4ST project funded by Programme Johannes Amos Commenius, call Excellent Research, (Project No. CZ.02.01.01/00/22 008/0004572).
Optical and magnetic properties of MXene-based quantum dots
Call: 30th Open Access Grant Competition; OPEN-30-18
Researcher: Barbora Vénosová
Institution: University of Ostrava
Field: Material Sciences
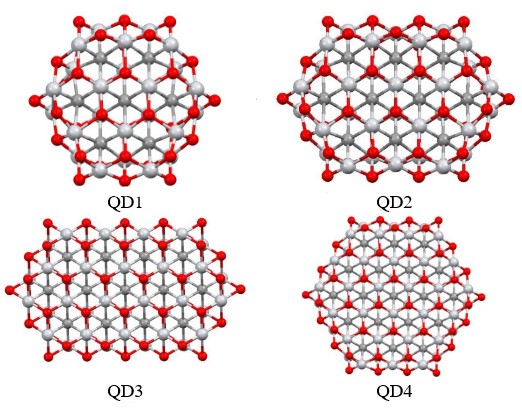
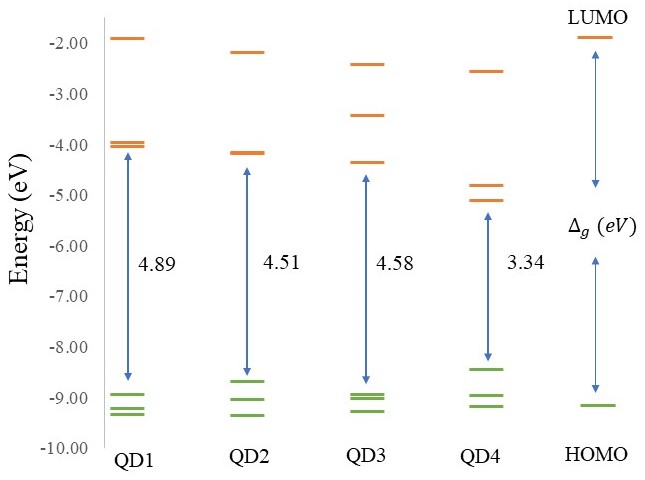
MXenes have attracted a lot of interest in the world of 2D materials due to their unique properties. MXenes are composed of transition metal atoms (M = Ti, V, Sc, Mo or Nb), carbon or nitrogen atoms (X), and terminal surface groups (T = -O, -OH, -F, -Cl and others) and have the general formula Mn+1XnTx. Due to the variety of M and X elements and the differences in surface and edge terminations, thousands of different MXene structures with unique properties can be prepared. For this reason, MXenes have become a rapidly expanding family of 2D materials. MXenes, with their characteristic planar structure, exhibit exceptional structural stability and good electrical conductivity. Their surfaces are tunable, and they possess a host of unique chemical properties. These features make them versatile, finding uses across a broad spectrum of applications. From biosensors and batteries to 10 nanometers in size) could improve or even create new properties. This is achieved by a combination of edge effects, surface, and quantum confinement.
The goal of our project is to control the tuning of MXene quantum dots (MXQDs) properties by modeling their size and structure. The first results show that the electronic and magnetic properties are strongly dependent on the size of the quantum dots. Based on these results, we want to investigate the optical and magnetic properties of different types of MXQDs in detail. First, we want to investigate the influence of surface functional groups on the optical properties using time-dependent density functional theory (TD-DFT). Subsequently, systematically study the different modeling options of MXQDs (different combinations of M, and X atoms with different functional groups) to tune the electronic, optical, and magnetic properties. This could provide opportunities for the preparation of new MXQDs structures with desired properties in many application areas (e.g. photonics, optoelectronics, and/or photocatalysts).
This research is supported by the European Union under the LERCO project (number CZ.10.03.01/00/22_003/0000003) via the Operational Programme Just Transition.
Towards sub-chemical accuracy of ab initio atomistic simulations of fusion enthalpies for molecular crystals
Call: 30th Open Access Grant Competition; OPEN-30-57
Researcher: Ctirad Červinka
Institution: University of Chemistry and Technology, Prague
Field: Material Sciences
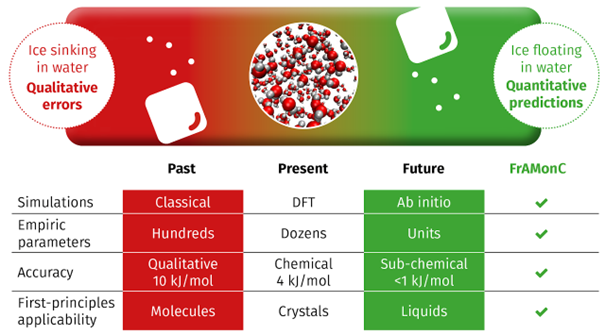
Organic molecular materials are ubiquitous, spanning diverse areas from pharmaceuticals, over fertilizers, explosives to semiconductors. Fabrication of final products in those fields often requires a sufficient solubility of relevant precursors, and subsequently, availability of a viable crystallization scheme of the target compound from solution. Understanding the melting and crystallization processes of such materials is important to design novel efficient fabrication techniques and to localize the limits of operation windows, e. g. temperatures and pressures at which a target material maintains its desired properties.
Computational modelling of crystal structures and their melting with a full resolution of individual atoms can significantly contribute to this understanding and it goes hand in hand with our capabilities of predicting material solubility, thermal stability and other important properties. Existing protocols based on classical molecular-dynamics simulations fail to reach the chemical accuracy of such predictions, and furthermore, they offer only a limited predictive power. Development of accurate and widely transferable quantum-chemical models of the melting parameters for molecular materials will thus significantly contribute to the material research.
Current project exploits the computational capacity of the IT4Innovations infrastructure to search for optimal ways of incorporating modern theoretical models to describe mutual interactions of molecules in bulk solid or liquid materials.
Our aim is to establish a high-throughput computational methodology, being sufficiently accurate at the same time, for an initial screening of material properties. In future, it should be performed at the initial stage of material research for a vast number of candidate materials, enabling to preselect the most promising targets exhibiting the properties desired for a particular application (e. g. solubility of drugs, or thermal stability of semiconductors). Only then, a detailed experimental refinement and verification narrowed only to the most promising targets could follow, which would render material research more efficient by circumventing large portions of experimental work that can be very costly and labor-demanding.
Structure Elucidation of Cu2O Photocatalyst Surfaces via Machine Learning
Call: 29th Open Access Grant Competition; OPEN-29-33
Researcher: Christopher Heard
Institution: Charles University in Prague
Field: Material Sciences
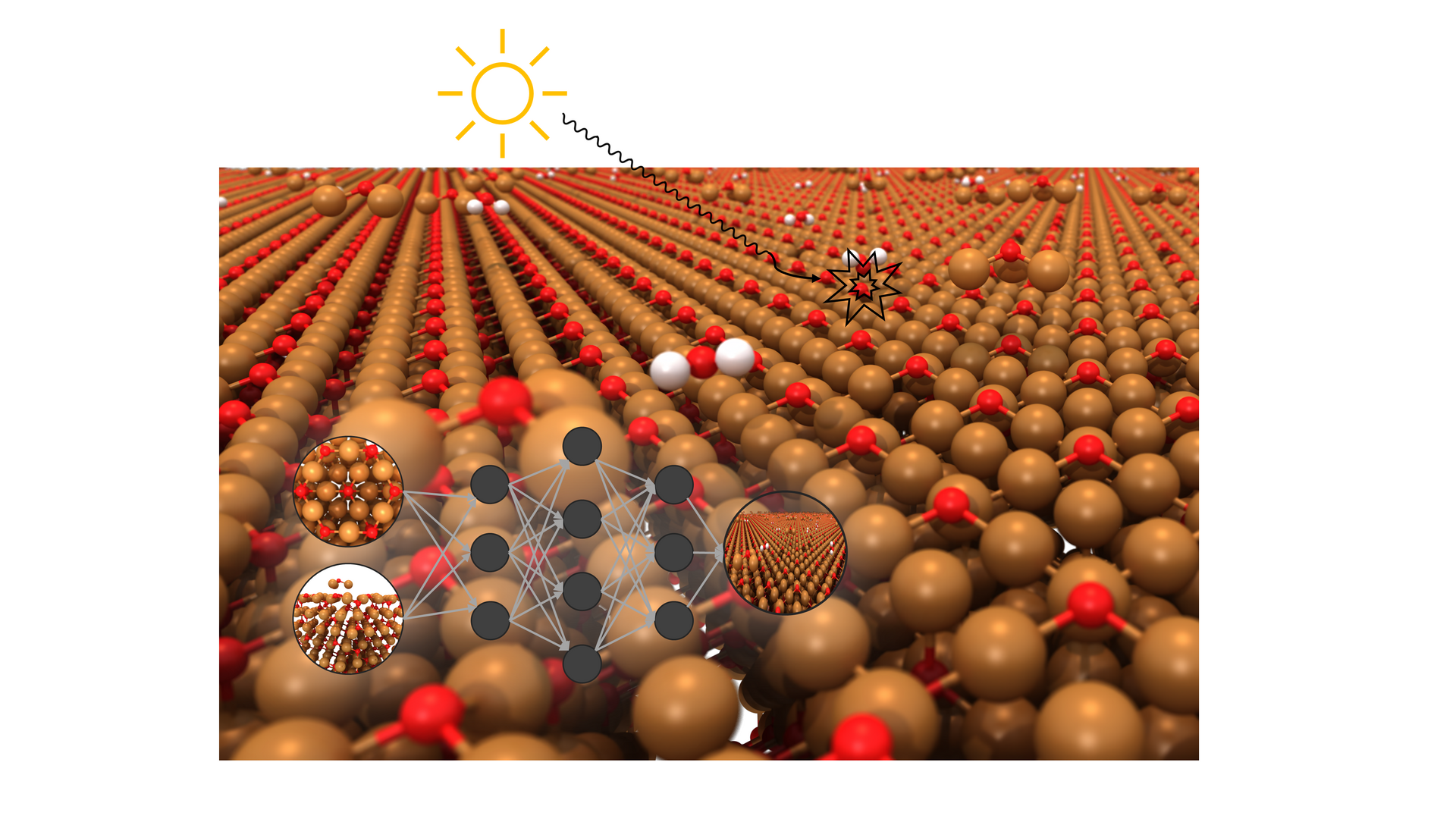
Cuprous oxide (Cu2O) is a material with promising applications in photo(electro)catalysis, green fuels, and solar energy production. However, its performance depends on the structure of its surface, which can change under different conditions, such as temperature, pressure and the presence of various naturally occurring adsorbate molecules. To elucidate the atomic-level morphology of Cu2O surfaces and thus understand its reactive properties, advanced simulation techniques are required, which are able to span the complexity and size of the system adequately.
This project involves the development and application of machine learning potentials (MLPs) for fast and accurate modelling of the structure and dynamic behaviour of Cu2O. It will leverage the computational power of supercomputers to generate a sufficiently large and reliable dataset of surface structures and temperature-dependent surface dynamics. The MLPs will be trained via quantum chemical calculations and validated against both high-level calculations and state-of-the-art experimental data and applied to understanding the atomistic nature of this promising catalyst.
The use of computationally expensive DFT functionals for salt-cocrystal systems investigation
Call: 29th Open Access Grant Competition; OPEN-29-25
Researcher: Simona Chalupná
Institution: University of Chemistry and Technology, Prague
Field: Material Sciences
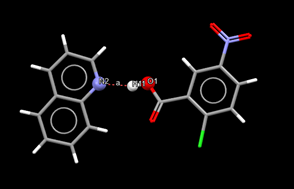
There is currently a wide range of different solid forms of pharmaceutical substances. One of these forms are pharmaceutical salts, which are commonly used to formulate active pharmaceutical ingredients (APIs). Approximately half of the drugs found on the market today are in the form of salts. Another solid form that is increasingly used in the formulation of pharmaceuticals is a cocrystal. The difference between these two forms is due to the position of only one atom - hydrogen. The exact position of this hydrogen, and hence the type of solid form, is crucial, as pharmaceutical companies must disclose it in patent and registration documentation, as required by medical authorities. Since the difference between the salt and the cocrystal is so small, it is interesting to conduct research into new methods of identifying the position of hydrogen.
This project deals with the possibility of distinguishing between salts and cocrystals by means of a calculation based on electron density functional theory (DFT). DFT describes electrons as glue poured between atomic nuclei. A functional is a mathematical function that describes the behaviour of such glue. The functionality and accuracy of this computational solution must be tested on a large number of substances (on the order of hundreds of structures), and subsequently, it is necessary to deal with those structures where the calculation does not agree with the experimental results. Thanks to IT4Innovations' allocated computational resources, this task can be performed using state-of-the-art methods, even if they are extremely computationally expensive.
Exploring the Intrinsically Disordered Domains in p53 Protein
Call: 29th Open Access Grant Competition; OPEN-29-55
Researcher: Amina Gaffour
Institution: Charles University, Faculty of Pharmacy in Hradec Králové
Field: Material Sciences
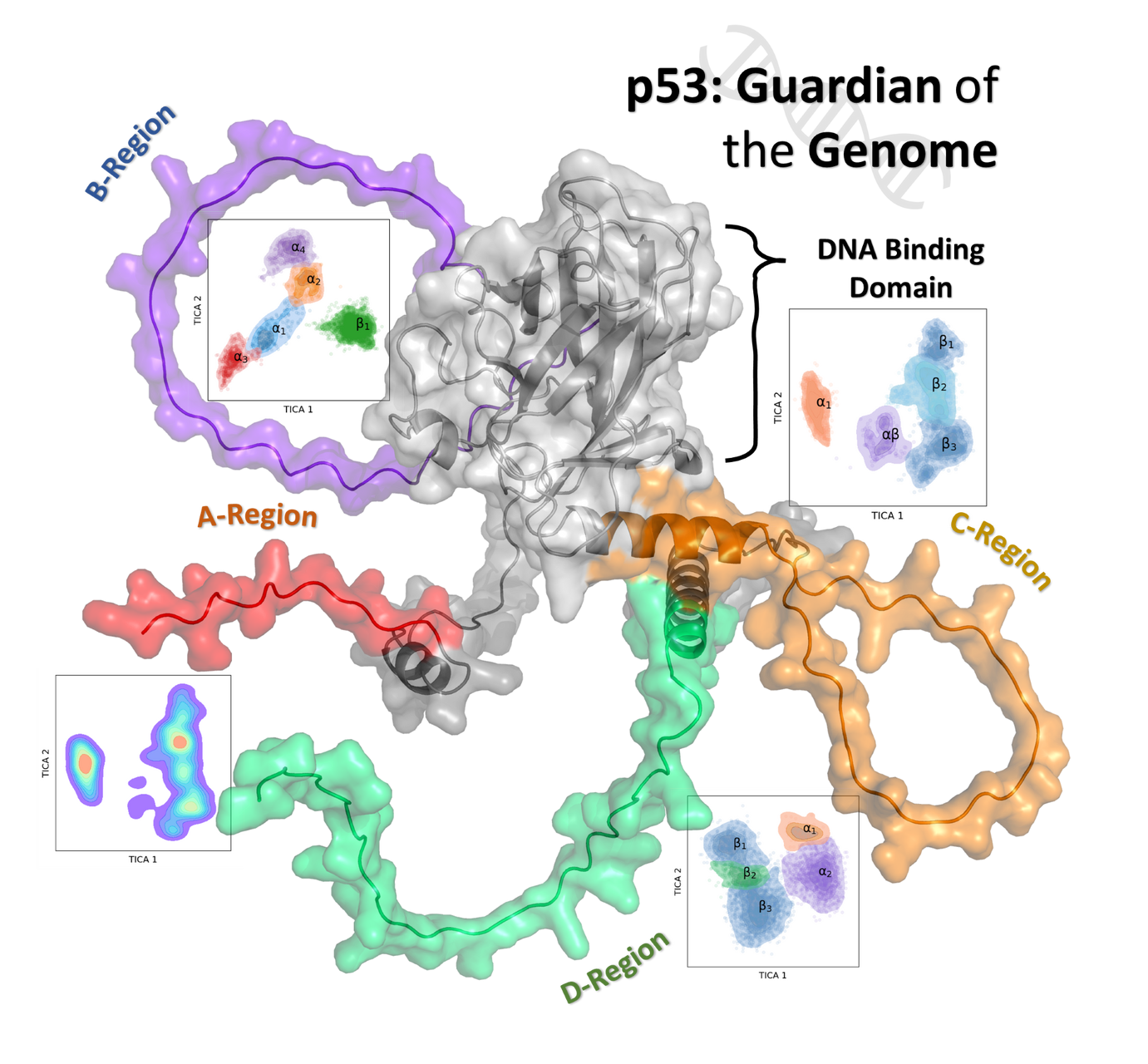
The p53 tumour suppressor protein is a critical cell cycle arrest and apoptosis regulator. Unfortunately, the molecular mechanisms underlying p53 function are not fully understood, particularly in intrinsically disordered regions (IDR), which have been observed to play an essential role in the protein’s folding, stability, and, most importantly, function. In this study, we will implement molecular dynamics to investigate the IDRs in p53, focusing on phosphorylation sites, ion concentration, the tetramerization process on secondary structure, and the function of the tumour suppressor protein. Preliminary findings are promising, showing a well-established effect on small trajectories from the concentration of ions and an alteration in how phosphorylated residues behave. Our findings will provide new insight into the protein's inner workings, which may provide key information for developing new therapeutic strategies for treatment.
Investigation of stability-activity relationship of transition metal-doped defective graphene for selected catalytic reactions
Call: 28th Open Access Grant Competition; OPEN-28-56
Researcher: Rostislav Langer
Institution: IT4Innovations
Field: Material Sciences
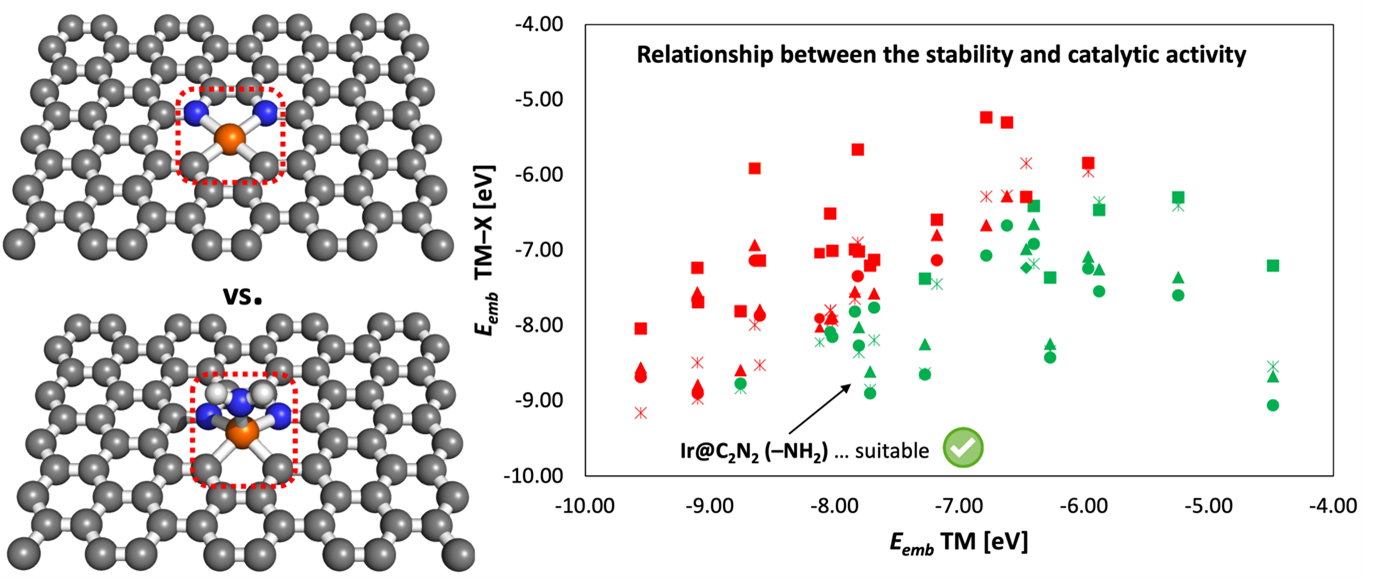
"Single atom catalysis" is a type of heterogeneous catalysis in which metal atoms are dispersed as single atoms on the surface of the support material. It has gained considerable attention in recent years due to its potential advantages over traditional catalysts, such as increased catalytic activity, better stability, and generally lower costs. This research project aims to investigate the stability and efficiency of single-atom catalysts anchored on 2D materials and understand the relationship between the stability of the active site and the efficiency of selected catalytic reactions. Density functional theory (DFT) is used for the calculations using finite and infinite models with periodic boundary conditions. The results of this research will significantly impact the development of more efficient and cost-effective catalysts for various industrial applications.
Organic molecules with inverted singlet-triplet gaps
Call: 28th Open Access Grant Competition; OPEN-28-50
Researcher: Libor Veis
Institution: J. Heyrovsky Institute of Physical Chemistry of the Czech Academy of Sciences
Field: Material Sciences
Organic electroluminescent diodes (OLEDs) form the basis of modern digital displays. To maximise their efficiency, they are made up of molecules with a small energy gap between the non-luminous triplet and luminous singlet excited states. The latest design of OLED materials (5th generation) features molecules with a negative energy gap, i.e., the excited singlet state has energy lower than the triplet state (INVEST molecules). In this case, the efficiency can reach
100 %.
The project aims to test the accuracy of our developed computational methods for adiabatic coupling on known INVEST molecules and find an optimal computational protocol for searching new 5th-generation OLED material candidates. The research is funded by the Grant Agency of the Czech Republic (GACR project No. 22-04302L).
Accuracy and precision for extended systems X
Call: 27th Open Access Grant Competition; OPEN-27-22
Researcher: Jiří Klimeš
Institution: Charles University in Prague
Field: Material Sciences
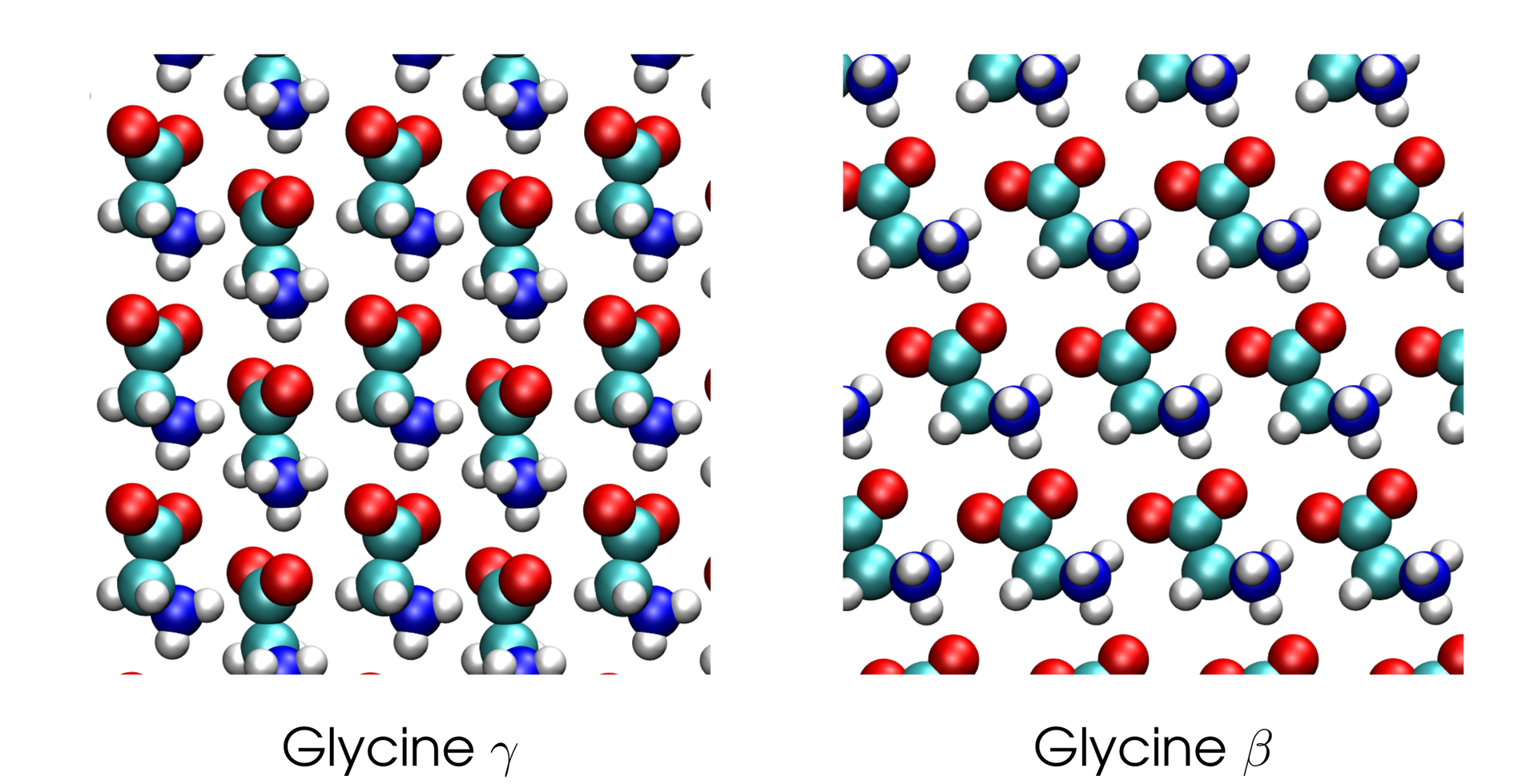
Many molecules can crystallise in different crystal structures, called polymorphs. For example, glycine can form β a γ structures, shown in the figure. It is important to know the polymorphs of pharmaceutically active molecules to avoid crystallisation in an unwanted structure. Computational methods based on quantum mechanics have been very useful to determine the structures and energies of possible polymorphs.
Within the project, we will consider two aspects of crystal energy calculations. First, we will test a method we have developed to reduce the error caused by neglecting core electrons in the calculations. Second, we will test the effect of different numerical parameters on the calculation of polymorph energies. The data obtained will lead to achieving higher reliability of crystal energy calculations and reduced computational time required to obtain the results.
Our long-term work goal is to develop methods for reliable predictions of the cohesive properties of molecular solids. Our research is supported by ERC within the APES grant (Accuracy and Precision for molecular solids, Horizon 2020 No. 729721).
Interplay between magnetism and superconductivity in the U-Te system under extreme conditions
Call: 27th Open Access Grant Competition; OPEN-27-37
Researcher: Urszula D. Wdowik
Institution: IT4Innovations
Field: Materials Science
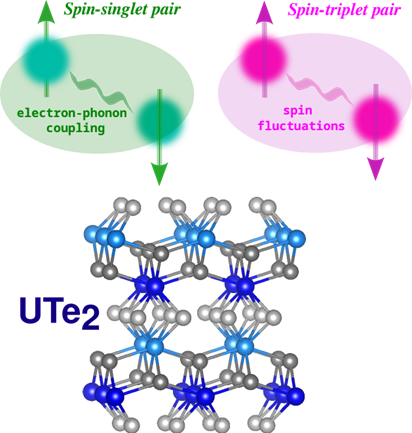
The interplay between magnetic fluctuations and superconductivity is one of the central issues in unconventional superconductors such as iron-based pnictides/chalcogenides, high-TC cuprates, and newly discovered uranium-based heavy-fermion systems featuring signatures of the spin-triplet Cooper pairing mechanism. Indeed, compounds belonging to the U-Te system that exhibit both strong electron correlations and magnetism are considered as promising candidates for realizing the chiral-triplet topological superconductivity, and hence they are materials of interest in quantum computing. Their unique properties are expected to be dramatically changed by external pressure and magnetic field.
The present research addresses exploration and understanding of the unconventional superconducting behaviour under external pressure of some selected binary compounds from the U-Te phase-space. The results of this project can deliver basic knowledge relevant also for other materials with unconventional types of superconducting pairing and topological superconductivity, which can find application in future quantum technologies.
This research will support the ongoing experimental studies performed within the Czech Science Foundation (GAČR) project No. 22-22322S entitled Unconventional superconductors under extreme conditions (co-PI Dr Dominik Legut, IT4Innovations).
Boron-containing catalysts for alkanes oxidative dehydrogenation
Call: 26th Open Access Grant Competition; OPEN-26-42
Researcher: Ota Bludský
Institution: Institute of Organic Chemistry and Biochemistry
of the Czech Academy of Sciences
Field: Material Sciences

Light olefins (alkenes) are important building blocks of the chemical industry as they are raw materials for the production of polymers, fuel additives, and other important substances. Their global consumption is steadily increasing. In 2019, ethylene and propylene consumption reached 160 and 115 million tonnes, respectively, and is expected to grow by 3.5-4% year-on-year over the next decade. The main objective of this project is to understand the nature of the active sites in boron-based catalysts for the selective oxidative dehydrogenation (ODH) of light alkanes (ethane, propane) to the corresponding olefins. The combination of experimental and theoretical approaches is essential for a deeper analysis of the observed measurements. The IT4I infrastructure plays an important role in theoretical calculations where conventional computers are not sufficient in capacity (ab initio molecular dynamics).
Machine learning for vacancy formation probability prediction in nitrogen-doped graphene – data collection for algorithm training
Call: 26th Open Access Grant Competition; OPEN-26-38
Researcher: Dagmar Zaoralová
Institution: IT4Innovations
Oblast: Material Sciences

Nitrogen-doped graphenes (NG) are very interesting materials that are applicable in many research domains. Previous studies have shown that the properties of NG depend on the amount of nitrogen embedded and the number of defects in the crystal lattice. Therefore, by fine-tuning the atomic structure of NG we could obtain materials with properties desirable for specific applications. However, it is almost impossible to manually design all possible forms of the NG atomic structure. Fortunately, machine learning approaches offer ways to address this problem. In order to train a sufficiently accurate algorithm capable of predicting the structure and properties of NG, it is necessary to collect sufficient information about NG with different atomic structures, which we plan to do through Density Functional Theory (DFT)-based calculations. We believe that the obtained algorithm will help in selecting probable NG structures that are interesting for further applications.
Thermopower in superconducting nanohybrids
Call: 25th Open Access Grant Competition; OPEN-25-41
Researcher: Vladislav Pokorný
Institution: The Institute of Physics of the Czech Academy of Sciences
Field: Material Sciences
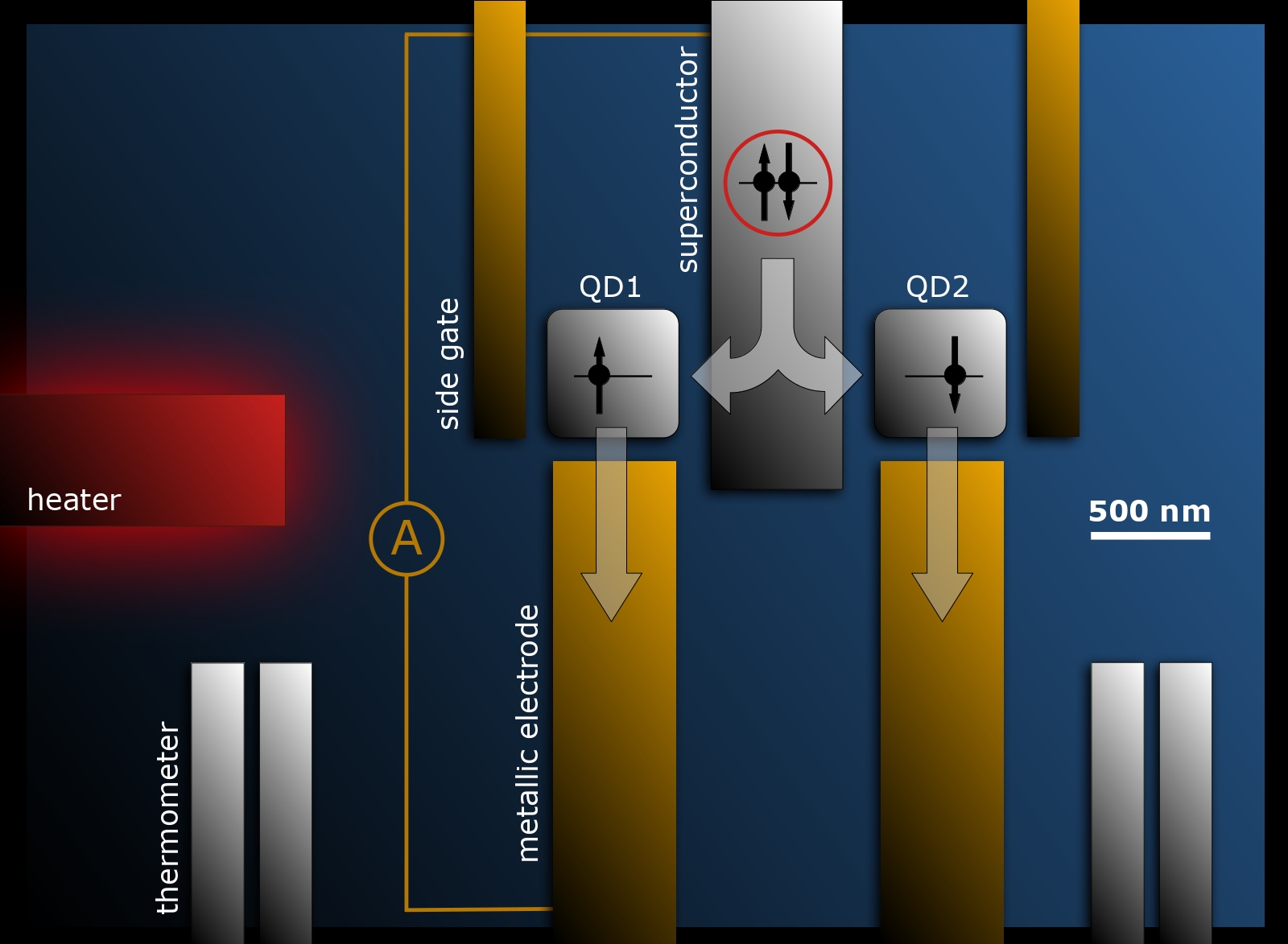
Nanoscopic devices consisting of quantum dots connected to superconducting electrodes are objects of study for a long time due to their applications in quantum computing. The advancements in experimental methods lead to the existence of increasingly complex structures and this is followed by the development of theoretical computational methods to describe their behaviour.
The topic of this project is the theoretical study of such devices consisting of a pair of quantum dots connected to superconducting and metallic electrodes. Their importance lies in their capability to create so-called entangled electron pairs with applications in quantum computing. One possibility of how to generate such pairs is the thermoelectric effect, in which the current is generated by the difference between the temperatures of the electrodes. The aim of this project is to simulate this behaviour using the quantum Monte Carlo method, which can reliably describe the important effects of electron correlations.
Physics of MXenes – Promising 2D Materials
Call: 24th Open Access Grant Competition; OPEN-24-46, multiyear
Researcher: František Karlický
Institution: University of Ostrava
Field: Material Sciences
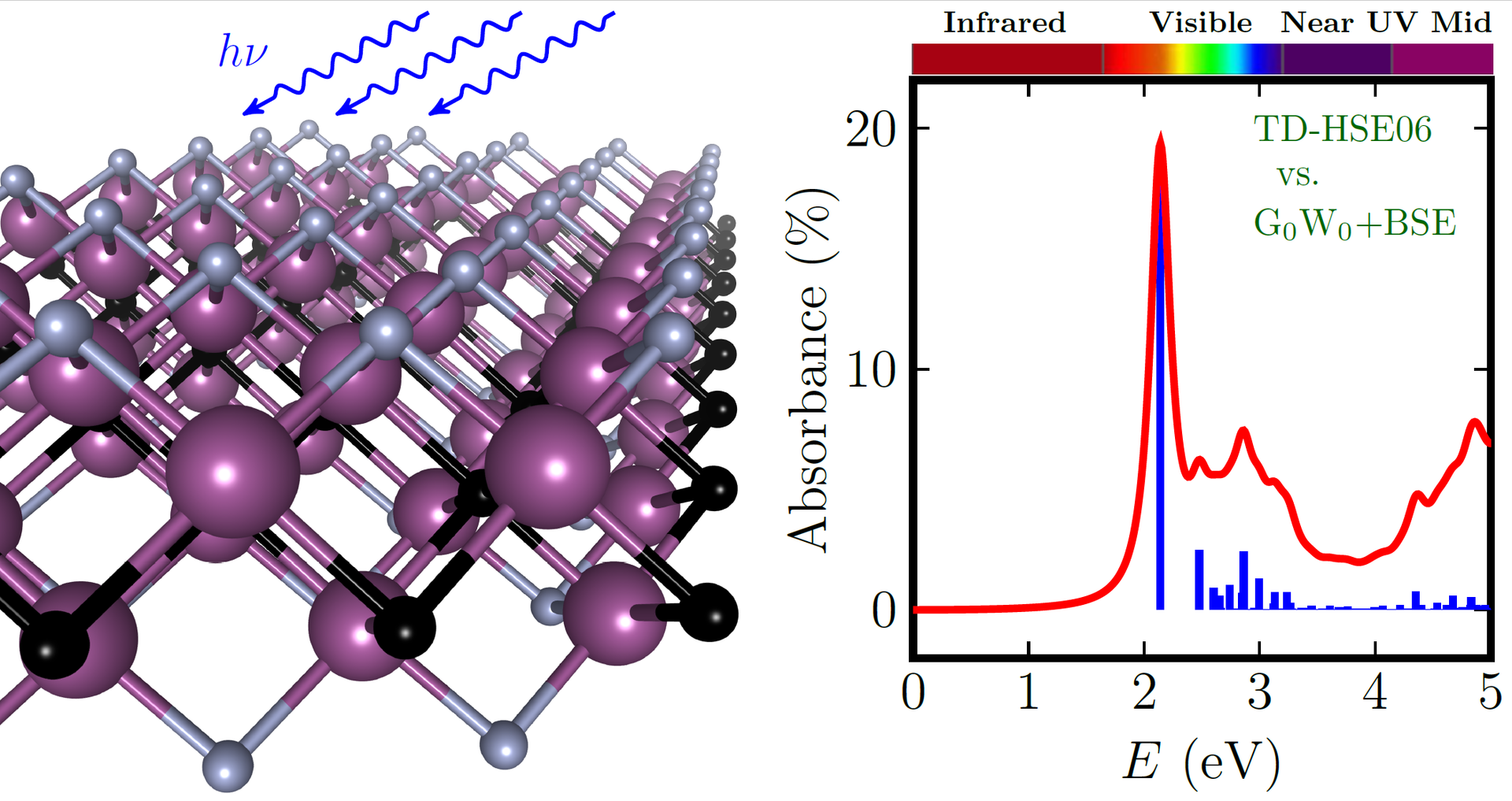
MXenes, i.e. transition metal carbides or nitrides, are relatively recently discovered two-dimensional (2D) materials. These materials are suitable candidates for numerous technological applications (electronics, photovoltaics) due to their durability and a wide range of physical properties (e.g. magnetic properties, variable bandgap, from metals to semiconductors), which is due to the variability of composition and the possibility of surface functionalization of MXenes. Both conducting and semiconducting MXenes can be combined into heterostructures, further expanding the possibilities of this promising class of materials. František Karlický's team, which has long been involved in computer modelling of 2D material properties, will gain fundamental physical insights into the behaviour and properties of MXenes that can be used in experiments and technological practice. Accurate predictions of the electronic and optical properties of MXenes in particular are challenging both because of the methods and the computational complexity.
Properties of Nanoparticles Intended for Medical Applications
Call: 23rd Open Access Grant Competition; OPEN-23-10, multiyear
Researcher: Martin Friák
Institution: the Czech Academy of Sciences
Field: Material Sciences

By reducing the volume of a solid below a certain limit, the solid changes its properties. This is called a dimensional phenomenon, and the particles of about the size of a tenth the thickness of a spider's fiber are called nanoparticles. Nanoparticles are widely used in medicine. Because of their small size, they can deliver medicine directly to the affected tissue, act against bacteria and viruses, and help in the examination and treatment of tumors. Magnetic nanoparticles have a special status as they can be controlled outside the body by an external magnetic field and used to heat a tumour to temperatures leading to its destruction. The properties of nanoparticles are closely related to their structure and knowledge of this relationship helps in their preparation. Within the project supported by GA CR, the relationship between structure and magnetic properties is studied, and experimental findings are combined with calculations performed at IT4I (Dr. M. Friák) so as to enable deeper understanding of these relationships.
Photoexcited electrons in the complex structures of carbon dots
Call: 22nd Open Access Grant Competition; OPEN-22-11 multiyear
Researcher: Michal Otyepka
Institution: Palacky University Olomouc
Field: Material Sciences
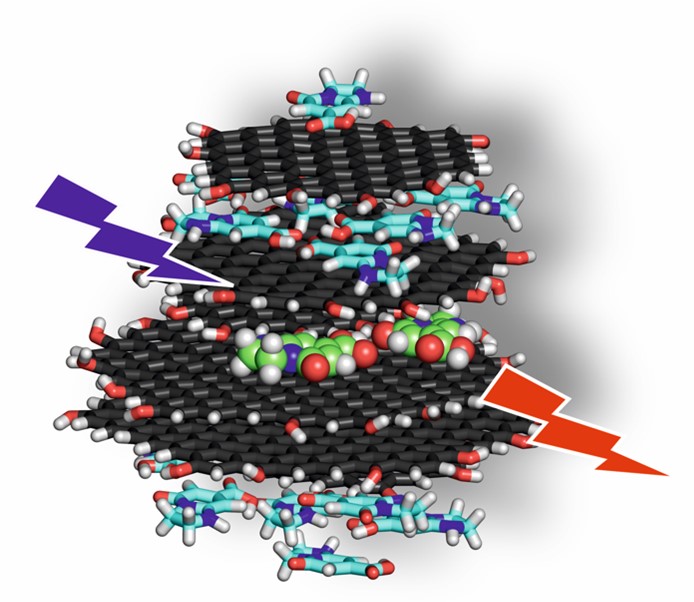
Carbon dots have only recently been discovered, yet today they represent one of the most intensively studied nanomaterials. Indeed, their excellent photoluminescence properties predestine them for a variety of applications ranging from LED light sources to biological imaging and medical diagnostics to chemical catalysis. This is also due to the fact that they are easy and cheap to prepare as well as they are stable and non-toxic. However, carbon dots are very reluctant to reveal their secrets, which makes it difficult to design them rationally and to optimise their properties in a targeted manner. Computer simulations are among the useful tools of the modern chemist, as they allow to answer questions that are very complicated for experiments. Simulations have been used for several years to understand the behaviour of carbon dots by the group led by Michal Otyepka. Thanks to the computational resources they have gained on IT4I computers, they will be able to study the mechanism by which a carbon dot absorbs light radiation, how the absorbed energy propagates through the dot structure, and how and why it is emitted. The scientists anticipate that if they understand this mechanism, it will allow them to target the design of carbon dots and expand their applications.
Calculation of magnetostriction via computational high-throughput approach
Call: 22nd Open Access Grant Competition; OPEN-22-10
Researcher: Pablo Nieves
Institution: IT4Innovations, VŠB-TUO
Field: Material Science
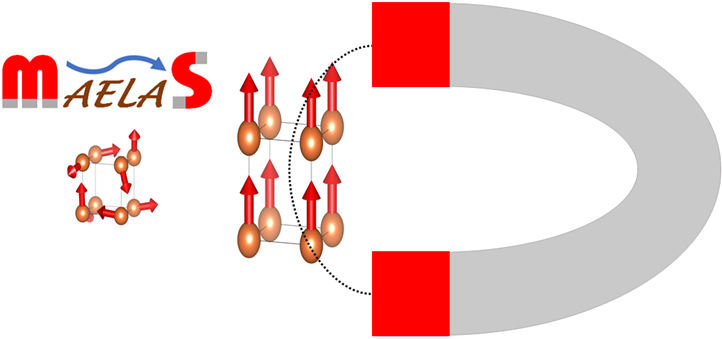
Magnetostriction is a physical phenomenon in which the process of magnetization induces a change in the shape or dimension of a magnetic material. Magnetostrictive materials are widely used in many technological applications like sensors (torque sensors, motion and position sensors, force and stress sensors) and actuators (sonar transducers, linear motors, rotational motors, and hybrid magnetostrictive/piezoelectric devices) where a high magnetostriction is required. Recently, we developed the program MAELAS [(version 1.0) P. Nieves et al., Computer Physics Communications 264 (2021) 107964, (version 2.0) P. Nieves et al., Computer Physics Communications (2021) 108197] to calculate magnetostrictive coefficients in an automated way. Presently, we are using MAELAS in IT4Innovations supercomputers to search for novel magnetic materials with high magnetostriction.
Thermal properties of Cerium Titanides
Call: 22nd Open Access Grant Competition; OPEN-22-18
Researcher: Andrzej Kadzielawa
Institution: IT4Innovations, VŠB-TUO
Field: Material Sciences
Our industry is still based on steam. In coal, gas, and nuclear power plants, the water produces electricity, the fuel used to heat it up. The reader, an astute person obviously, already sees significant problems: (i) carbon emissions; (ii) safety. Whilst emitting almost no CO2, nuclear energy has regrettably become loathsome after the Chornobyl and Fukushima incidents. Although the design of reactors based on fission (splitting atom nuclei into two or more particles, releasing excess energy) was significantly improved, there is another way. A clean way: Thermonuclear Fusion Reactors. The idea is simple: to reproduce the same process that a star does to produce energy. Fusion of two Hydrogen (H) atoms into Helium (He), c.f. [1].
In other words, we heat our steam engine with a small, artificial sun. Some of these devices are already operating (one in Prague [2]), yet there are still many engineering problems to solve. One of them is siphoning the radiating heat of plasma (temperature ~200 000 000 ℃, confined in the strong magnetic field) to water, i.e., a wall. While there is no possibility of an ecological catastrophe, the device itself can be damaged in an accident. Thus, the material for the reactor wall must withstand the so-called fail-safe scenarios.Trivializing, we have to consider the bombardment of our material with ions, atoms, and molecules of hydrogen (and its isotopes). Horse sense tells us that the wall should be hard, the classical physics, that the atoms it is built from should be heavy. These points provide a starting point: Tungsten (W) - a heavy yet stable and inexpensive element with high hardness. Unfortunately, while the performance in a vacuum is excellent, Tungsten is explosively oxidizing when exposed to the air. Therefore, coating the wall is out of the question, as a film might disrupt the heat conductivity can be easily damaged by plasma.
The next logical step is to create an alloy of Tungsten with a small addition of an element that (i) has an oxide that does not allow oxygen molecules to pass (making a make-shift, self-healing coating); (ii) is oxidizing at least as fast as Tungsten. Conveniently, adding ~10 % of Chromium (Cr) does precisely that. The story does not end here, as there is a new problem - W-Cr alloys slowly but steadily diffuse into a mixture of two: W- and Cr-rich grains. Our work is to find a third stabilizing element (X) of the W-Cr-X alloy slowing (or ideally stoping) the diffusion.
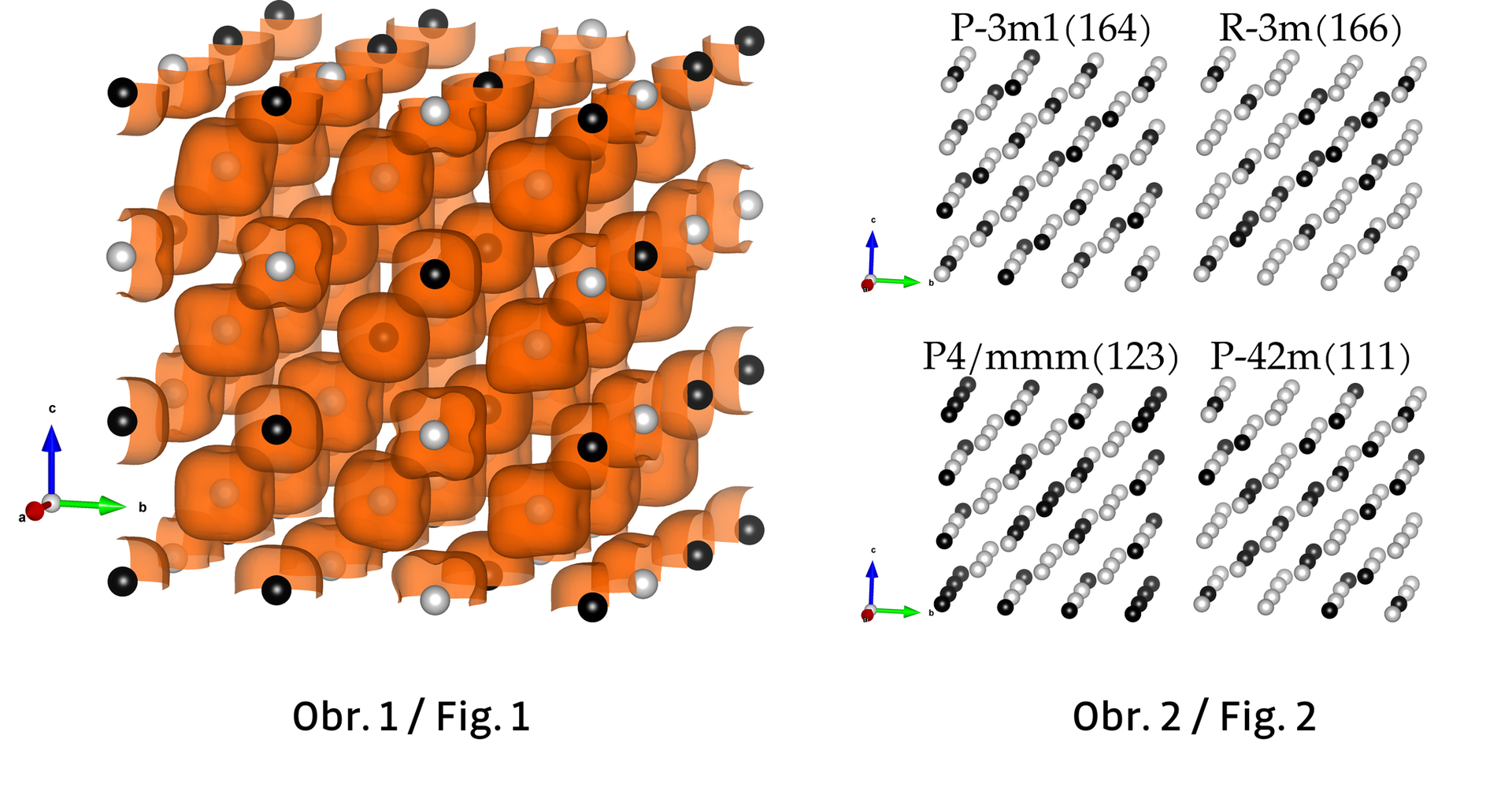
The first step of computational modelling is to reproduce the problem. The approach we chose is to describe an alloy as a quantum-mechanical system of electrons and ions, then use the outcome to build a statistical model. First, we create our building blocks using the Density Functional Theory (DFT [3,4]) by calculating the electronic behaviour on the lattice of Tungsten and Chromium atoms. The outcome charge distribution (Fig. 1) allows us to understand the electric and heat conductivities change with composition and the impact of lattice shape on vibrations of the ions in the lattice. We first use the IT4Innovations clusters to generate the Special Quasirandom Structures [6] (Fig. 2): the distributions of atoms in a finite cell, reproducing the alloy randomness in the best possible way. Then, the bulk of calculations occurs: we obtain each cell model's electronic and dynamic behaviour using the Vienna Ab initio Simulation Package (VASP [7]) by minimizing its energy (E). Then, having a representative set of states (a part of the statistical ensemble - set of all possible realizations), we can incorporate temperature (T). Intuitively it is not a big deal, but in reality, the temperature is anything but ostensible. In physics, it is a single parameter that describes the kinetics of the elements in a model (e.g., the average kinetic energy of gas particles). While it is rather inconvenient in understanding, it is useful to treat temperature as an intensive variable (non depending on the number of particles) coupled with extensive entropy (S) - the measure of disorder in the system. What is and how to describe, calculate, and understand entropy is a topic on a separate article (or a book); let us assume now that we can calculate it for each of the states calculated using VASP. Here, minimizing the so-called Free energy (F = E - S×T), we find the optimal state of each composition of elements in each temperature.
Completing the procedure described in the paragraph above for different X in W-Cr-X gives us information on the stability and properties of these alloys and whether it makes sense to synthesize them in a lab. Following this approach, we recently published promising results on Tungsten - Chromium - Hafnium (Hf) systems [8].
This project is a part of a GAČR standard grant No. 20-18392S Tailoring thermal stability of W-Cr based alloys for fusion applications.
[1] wikipedia.org, Fusion power, https://en.wikipedia.org/wiki/Fusion_power, accessed 1 November 2021.
[2] COMPASS, Institute of Plasma Physics of the Czech Academy of Sciences, Prague, http://www.ipp.cas.cz/vedecka_struktura_ufp/tokamak/COMPASS
[3] P. Hohenberg, W. Kohn, Phys. Rev., 136 (3B), pp. B864-B871 (1964).
[4] W. Kohn, L.J. Sham, Phys. Rev., 140 (4A), pp. A1133-A1138 (1965).
[5] K. Momma and F. Izumi, J. Appl. Crystallogr. 44, 1272-1276 (2011).
[6] A. Zunger, et al., Phys. Rev. Lett., 65 (3), pp. 353-356 (1990).
[7] G. Kresse, J. Furthmüller, Phys. Rev. B 54 (16), pp. 11169-11186 (1996).
[8] J. Veverka, et al., Mat. Lett. 304, 130728 (2021).
EXTENSIVE PURSUIT OF SINGLET FISSION SENSITIZERS
Call: 21st Open Access Grant Competition
Researcher: Diego López-Carballeira
Institution: Faculty of Electrical Engineering, Czech Technical University in Prague
Field: Material Sciences
Singlet fission is a process that allows a molecule to generate two triplet excited states with the absorption of a single photon. This feature implies deep advantages for the generation of solar energy, and the scientific community diligently tried to get novel sensitizers that could boost the efficiency of the future photovoltaic devices.
Unfortunately, all those efforts have been futile, and the few molecules that have been demonstrated to show efficient singlet fission lack the photostability required to be applied in a real device. In order to provide novel candidates to be used as singlet fission sensitizers, our group uses high level quantum calculations for the screening of large databases. With the 6.9 million CPU hours provided by IT4Innovations it is possible to seek among the plethora of known molecules stored in databases, modeling their electronic structure and selecting those that are found to be compatible with the presence of singlet fission.
The project aims to provide a list of readily available compounds compatible with the singlet fission process, narrowing down the number of candidates to be tested by expensive theoretical and experimental approaches.
MOLECULAR MODELING AND DYNAMICS STUDY OF MATERIALS FOR USE IN CONTROLLED RELEASE FERTILIZERS
Call: 20th Open Access Grant Competition
Researcher: Doc. Václav Čuba
Institution: Czech Technical University in Prague
Field: Material Sciences
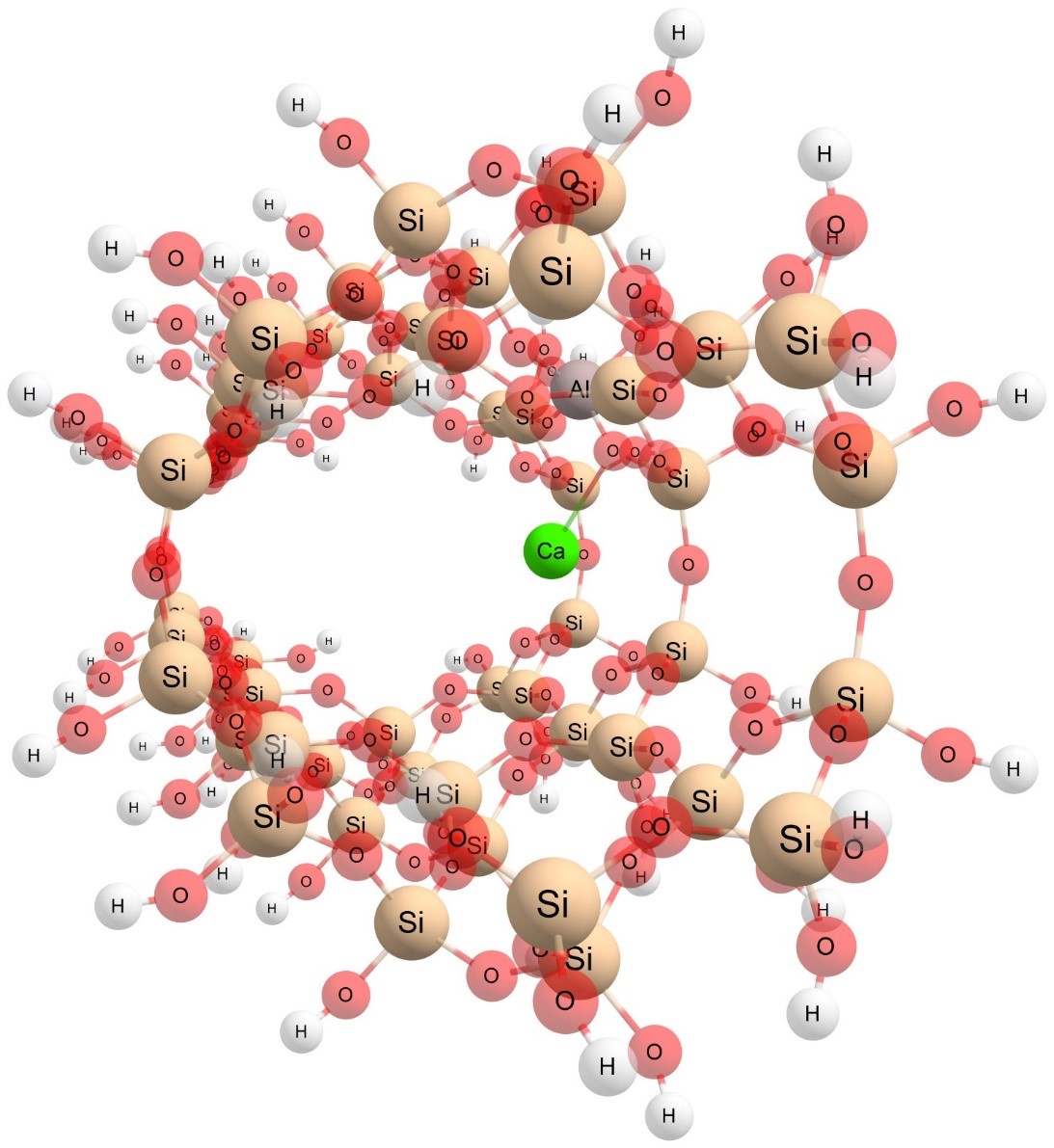
Introduction of NPP (nitrogen, phosphorus, and potassium) and other composite fertilizers made care of soil and plants much easier for farmers. These fertilizers increased yield but caused new and unforeseen ecological problems. To reduce environmental stresses connected mainly to an excess of nitrogen in soil and underground water, new slow and controlled release fertilizers, which copy the curve of plant nutrient uptake with their curve of nutrient release, were developed. This project is focused on finding and studying materials that are useable as a base for slow release fertilizers able to release nutrients in a controlled manner to the environment. Such promising materials seem to be zeolites, the properties of which allow the preparation of a fertilizer containing all essential nutrients, as well as having slow release properties. The binding of molecules of nutrients with zeolitic structures will be studied by molecular modeling methods. From the modeling results, it is possible to predict how the release of nutrients will take place, and what the difference in the strengths of the bonds between each nutrient and the zeolitic structure will be. The aim of the project is to study the possibilities of preparing slow release fertilizers.
SUBSTITUTED HYALURONAN MOLECULES IN AQUEOUS AND MIXED SOLVENTS
Call: 20th Open Access Grant Competition
Researcher: Dr Marek Ingr
Institution: Tomas Bata University in Zlín
Field: Material Sciences
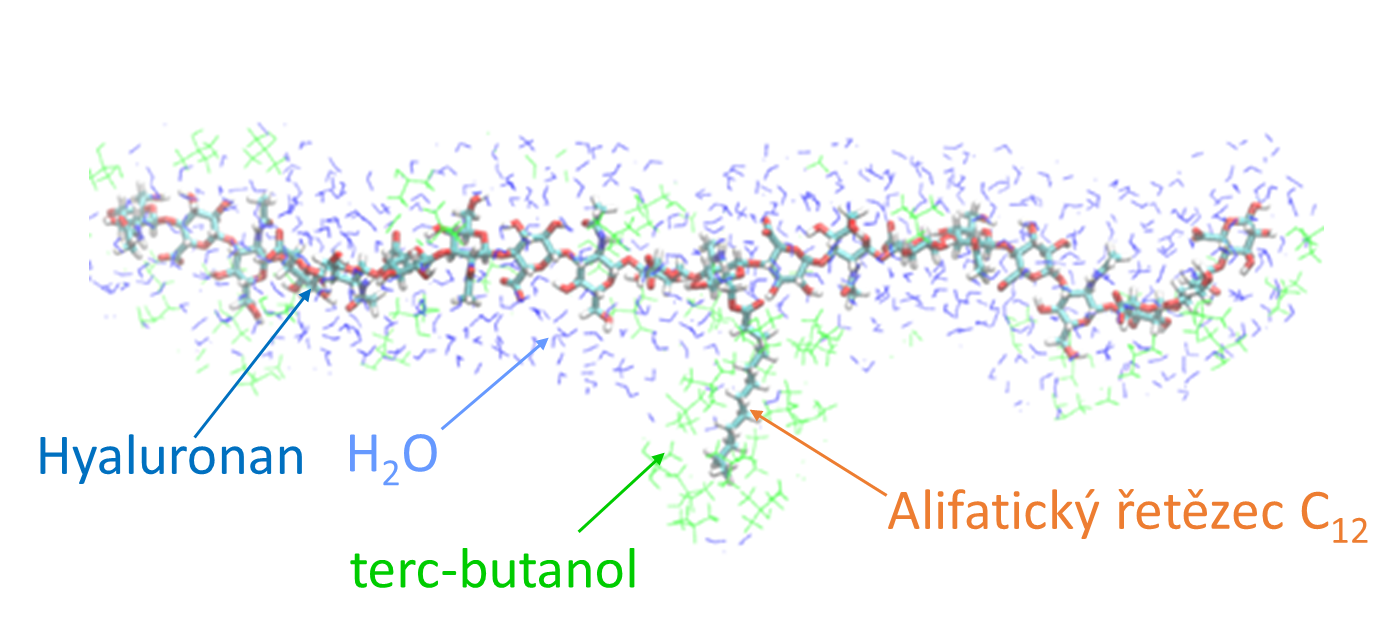
Hyaluronic acid (Hyaluronan, HA) is a key component of the extracellular matrix of skin and connective tissues. It finds wide applications in cosmetics and pharmacology as a compound supporting tissue regeneration and wound healing. As a biocompatible material, it is also used as a base for drugdelivery systems and tissue repair. Although HA is highly hydrophilic, many of its technological applications often require its use in non-aqueous environments. Following previous studies of free HA macromolecules in aqueous solutions and mixtures of water and miscible organic matter, Dr. Marek Ingr and his research team are now looking at the structure of HA molecules substituted by one or more aliphatic chains in the same solvents. The equilibrium behaviour of the molecules is simulated by the molecular dynamics method, from which the molecule‘s conformation and the mutual position and interaction of its parts, the separation of the mixed solvent around the individual parts of the molecule, the interaction of the parts of the molecules with the molecules in the solution, and in particular the precursors of subsequent substitution reactions, are evaluated. The results of these studies will contribute to optimizing responses leading to the formation of modified HA molecules and the design of materials based on them that can find applications in healthcare, cosmetics, and other fields.
DISCOVERY OF NOVEL EFFICIENT CATALYSTS FOR CO2 CAPTURE AND IN SITU UTILIZATION: DFT INVESTIGATION
Call: 20th Open Access Grant Competition
Researcher: Dr Valeria Butera
Institution: CEITEC, Brno University of Technology
Field: Material Sciences

Carbon dioxide (CO2) is considered the main cause of global warming. Over half of CO2 emissions are from large industrial point sources, while the remainder of these emissions are from small mobile sources. Reducing its emission below critical levels requires not only political commitment but also novel scientific approaches to capture CO2 and to enable its conversion from a waste product into value-added products. The key measure to achieve this will be efficient and practicable processes for ambient air and large-scale CO2 sequestration and utilization. The main focus of this project is the development of innovative technologies aimed at slowing or stemming anthropogenic carbon emissions. The challenge here is the design of new efficient, selective, and “green” homogeneous and heterogeneous catalysts for CO2 capture and CO2 conversion. Valeria Butera and her team will first focus on materials that are suitable for CO2 sequestration and conversion directly from ambient air (direct air capture, DAC). The results of this investigation will pave the way for developing suitable technologies to extract CO2 from large industrial sources.
DESIGN OF A NEW SMART MATERIAL WITH MAGNETIC SHAPE MEMORY EFFECT
Call: 19th Open Access Grant Competition
Researcher: Dr Martin Zeleny
Institution: Faculty of Mathematics and Physics, Charles University in Prague
Field: Material Sciences
Magnetic shape memory (MSM) alloys have a large application potential in actuators, sensors, energy harvesters, and magnetic refrigeration systems thanks to the extraordinary properties of their multiferroic martensite structure. The macroscopic deformation of such materials in an external magnetic field is caused by the motion of highly mobile twin boundaries in combination with high magnetic anisotropy. However, operating temperatures of currently used materials are too low for engineering applications, which is caused by their low transformation temperatures between austenite and martensite. Within the OP RDE MATFUN project, which was awarded more than 4.1 million core hours in the first period, Martin Zeleny will search for new materials with a high application potential that combine the stability of martensitic phase up to high temperature with its high magnetic anisotropy and low twining stress necessary for motion of twin boundaries, which are also necessary prerequisites of a successful MSM alloy development. Such alloys will in turn enable the miniaturisation and development of new devices in robotics, automotive, aerospace, and biomedical industries. In addition to finding new candidates for experimental preparation, Martin Zeleny will also investigate in depth the basic aspects of the multiferroic behaviour of alloys with magnetic shape memory, such as the physical origin of the martensitic transformation or the mobility of twin boundaries.
THERMODYNAMICS OF ACTINIUM METAL
Call: 19th Open Access Grant Competition
Researcher: Lukas Kyvala
Institution: IT4Innovations, VSB-TUO
Field: Material Sciences

The half-life of the most stable actinium isotope is only 21.77 years. Therefore, its concentration in nature is significantly low. Were it not for one of the products of the decay of thorium and uranium, it would have long since disappeared from Earth. Due to its high radioactivity, only a few experiments on metal actinium were done and actinium remains as one of the least explored naturally occurring elements. Even the basic property as lattice constant has been subject of discussion for many years and its unusually small value has not been fully elucidated. Lukas Kyvala will use the awarded computational resources (272 000 core hours) to analyse physical properties of actinium, its stability under different temperatures, and the relativistic effects as actinium, due to its high radioactivity (150 times higher than in the case of radium) has become a preferred element in radiotherapy. Study of actinium might not only help understand the physics of actinoids but also enable the obtained knowledge to be utilized in its applications such as the source of neutrons and geochemical indicator for deep circulation of sea water. Moreover, its ability to deliver stable amounts of heat is suitable for generating electricity in space where solar power is not available (i.e., for missions on the dark side of the moon).
ACCURACY LIMITS OF QUANTUM MONTE CARLO IN WEAK-INTERACTION LIMIT III
Call: 18th Open Access Grant Competition
Researcher: Dr Matúš Dubecký
Institution: University of Ostrava
Field: Material Sciences
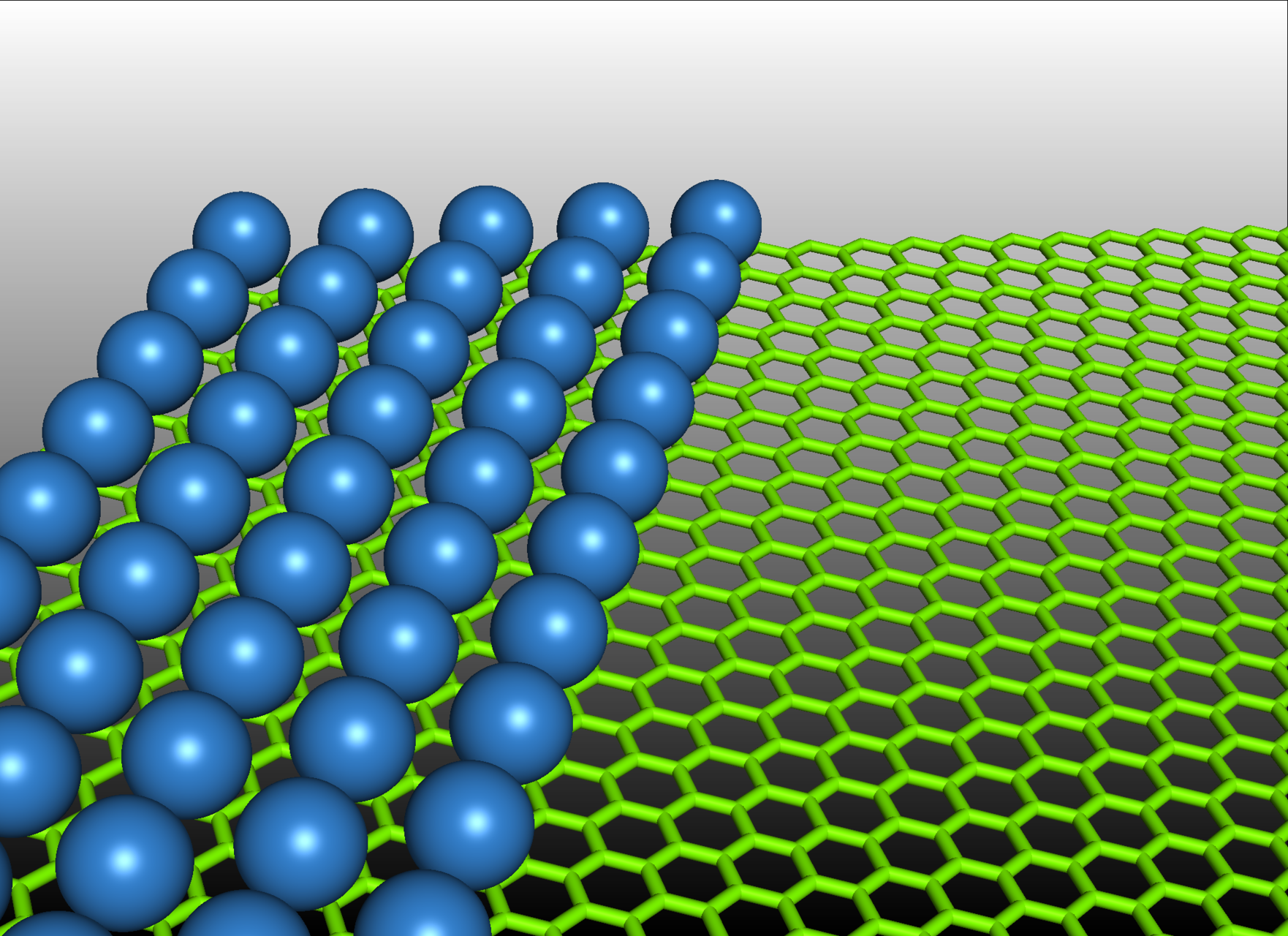
More than 4 million core hours were awarded to Matúš Dubecký for his research focused on determining the accuracy limits of the Fixed-node diffusion Monte Carlo (FNDMC) method for noncovalent interactions. Noncovalent interactions play a key role in many research areas such as material science and drug design. The team led by Matúš Dubecký will conduct a benchmark study as a follow up to their previous research and application of the FNDMC method, for example, in 2D materials, the properties of which are affected by noncovalent interactions of molecules, and 1D conductors on their surfaces. The objective of this project is to determine, by means of a supercomputer, the accuracy limits of the FNDMC method, which is currently frequently used as a quantum reference method for large noncovalent systems. Apart from gaining a physical insight into the FNDMC method and design of potential improvements, the results will lead to better accuracy control and more rational application of this method not only for large systems.
ACCURACY AND PRECISION FOR EXTENDED SYSTEMS IV
Call: 18th Open Access Grant Competition
Researcher: Dr Jiří Klimeš
Institution: Charles University in Prague
Field: Material Sciences

More than 2.8 million core hours of the IT4Innovations computational resources were awarded to a team led by Jiří Klimeš for a project focused on precision and accuracy of binding energies calculations in crystals, especially those bound by non-covalent interactions. These materials, such as methane clathrates at the bottom of the sea, pharmaceuticals crystals, and layered systems such as graphite to name but few. One of their peculiar properties is polymorphism – the ability of a crystalline material to adopt different crystal structures, even under same conditions. One of the objectives of this project is to use a supercomputer to develop a method that would allow a reliable description of the stability of different polymorphs or different crystalline phases of materials. It is a basic research project aiming at both gaining deeper understanding of the accuracy limits of the currently used methods and development of higher precision methods applicable in future material simulations. The research team led by Jiří Klimeš would also like to integrate developed scripts for preparation and analysis of calculation into “packages” used for automated working procedures. This all is expected to ensure that the methods for accurate calculations of binding energies can be used by other research groups as well as increase reproducibility of such results.
FROM ANTIPHASE BOUNDARIES TO NEW RARE-EARTH-FREE MAGNETS
Call: 17th Open Access Grant Competition
Researcher: Prof. Mojmír Šob
Institution: CEITEC
Field: Material Sciences

The research team led by Prof. Mojmír Šob from CEITEC was awarded more than 8 million core hours for the project focused on analysis of antiphase (AP) boundaries on magnetic properties of intermetallic compounds and their thermodynamic as well as mechanical stability. This information is essential for successful development of new magnetic materials. The project aims at Fe-Al alloys, the magnetic properties of which can be improved by AP boundaries by up to dozens of per cents according to the latest experiments. The awarded computational resources will be used by the research team to study the properties of conventional (rare-earth-free) Fe-Al-based magnets and to understand the relevant physical mechanisms, the knowledge of which is essential to improve the properties of these magnetic materials.
NEURAL NETWORK POTENTIALS FOR IN SILICO DESIGN OF ZEOLITES
Call: 17th Open Access Grant Competition
Researcher: Dr Lukáš Grajciar
Institution: Charles University in Prague
Field: Material Sciences
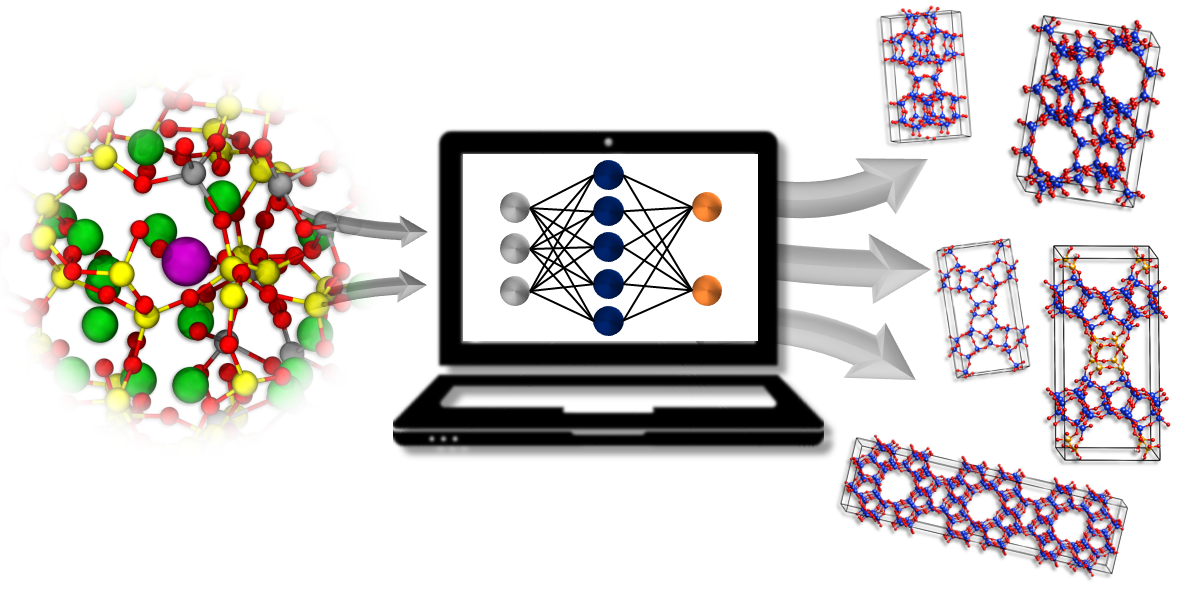
Lukáš Grajciar was awarded more than 2 million core hours for implementing his project focused on the in silico design of new catalysts, such as zeolites. Zeolites have great potential in the area of developing green technologies because they are the most important industrial catalysts used primarily in crude oil processing and petrochemistry. Lukáš Grajciar along with his colleagues Andreas Erlebach, Christopher J. Heard, and Petr Nachtigall use the awarded computational resources for simulations using deep neural network-based force fields for screening large databases of candidate structures and their modelling under operating conditions with unprecedented accuracy. The project results shall provide deeper insight into the structure and stability of existing and hypothetical zeolites, which have not yet been synthesized, and improve the catalytic properties of zeolites in general.
MOLECULAR AND MESOSCOPIC SIMULATIONS OF AQUEOUS SOLUTIONS IN INHOMOGENEOUS ENVIRONMENTS
Call: 16th Open Access Grant Competition
Researcher: Dr Barbora Planková
Institution: Czech Academy of Sciences
Field: Material Sciences
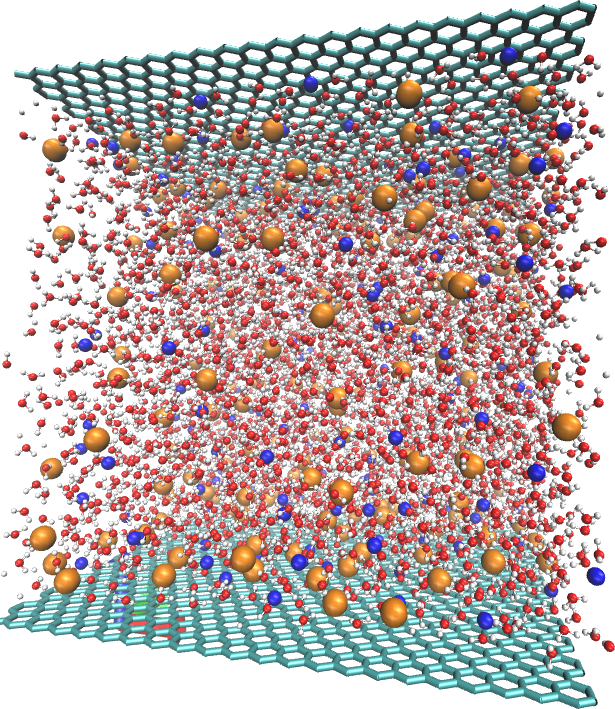
Barbora Planková from the Institute of Chemical Process Fundamentals of the Czech Academy of Sciences was awarded more than 1 million core hours for molecular and mesoscopic simulations of aqueous solutions in non-homogenous environments. Aqueous solutions are omnipresent in nature, industrial processes, and daily life. Understanding their behavior in inhomogeneous environments (nanopores, self-assembled systems) is important in many key applications such as medicine and enviromental protection. Together with her colleagues, Karel Šindelka and Martin Lísal, Planková will use the supercomputer in three research areas. The first one is graphene-aqueous electrolyte interfaces. Graphene is also called the miracle material of the 21st century. Graphene membranes, for example, could be used in water desalination or its cleaning. First of all, however, it is important to understand the elementary molecular processes, which will be studied by the project author using the supercomputer. The second research area is focused on ionic surfactants used, for example, in fabric conditioners. Part of the allocated computational resources will be used to study of the behaviour of these active substances and their interaction with soft surfaces – the key aspects of their functionality. The third research area focuses on solubilisation of small molecules in interpolyelectrolyte complexes, which can influence drug effectiveness and removal of pollutants.
HIGH-THROUGHPUT SCREENING OF METAL-ORGANIC FRAMEWORKS FOR CO2 SEPARATION FROM A POST-COMBUSTION GAS MIXTURE UNDER HUMID CONDITIONS
Call: 16th Open Access Grant Competition
Researcher: Dr Pezhman Zarabadi-Poor
Institution: CEITEC, Masaryk University
Field: Material Sciences
Dr. Pezhman Zarabdi-Poor from CEITEC was awarded more than 3 million core hours for identification of the best performing metal-organic frameworks that can separate CO2 from a post-combustion gas mixture using high-throughput systematic screening. The main anthropogenic source of CO2 emission is combustion of fossil fuels. Economic growth and industry development have continually been contributing to its concentration increase in the atmosphere, which leads to global warming of the Earth. One of the most efficient methods to avert this unwanted phenomenon and to maintain industrial development is Carbon Capture Sequestration (CCS). In this regard, metal-organic frameworks (MOFs) are considered attractive solid adsorbents that can efficiently be utilized for carbon capture from one of the main sources of CO2 emission, i.e. post-combustion gas (average content of CO2 is 15–16 %). The supercomputer and the computational resources in the amount of 3.3 million core hours will be used by Zarabdi-Poor to find best performing metal-organic frameworks, which will later be synthesized and experimentally verified in a laboratory. This research is part of the CMPSTORE project funded by the EU Horizon 2020 programme within the Marie Skłodowska-Curie action and co-funded by the South Moravian Region. The project is implemented within the research group of Prof. Radek Marek, and its active participant is Esmaiel Farajpour Bonab, a student of the Physical Chemistry PhD study programme.
IN SILICO DRUG DESIGN
Call: 14th Open Access Grant Competition
Researcher: Prof. Pavel Hobza
Institution: The Czech Academy of Sciences
Field: Material Sciences
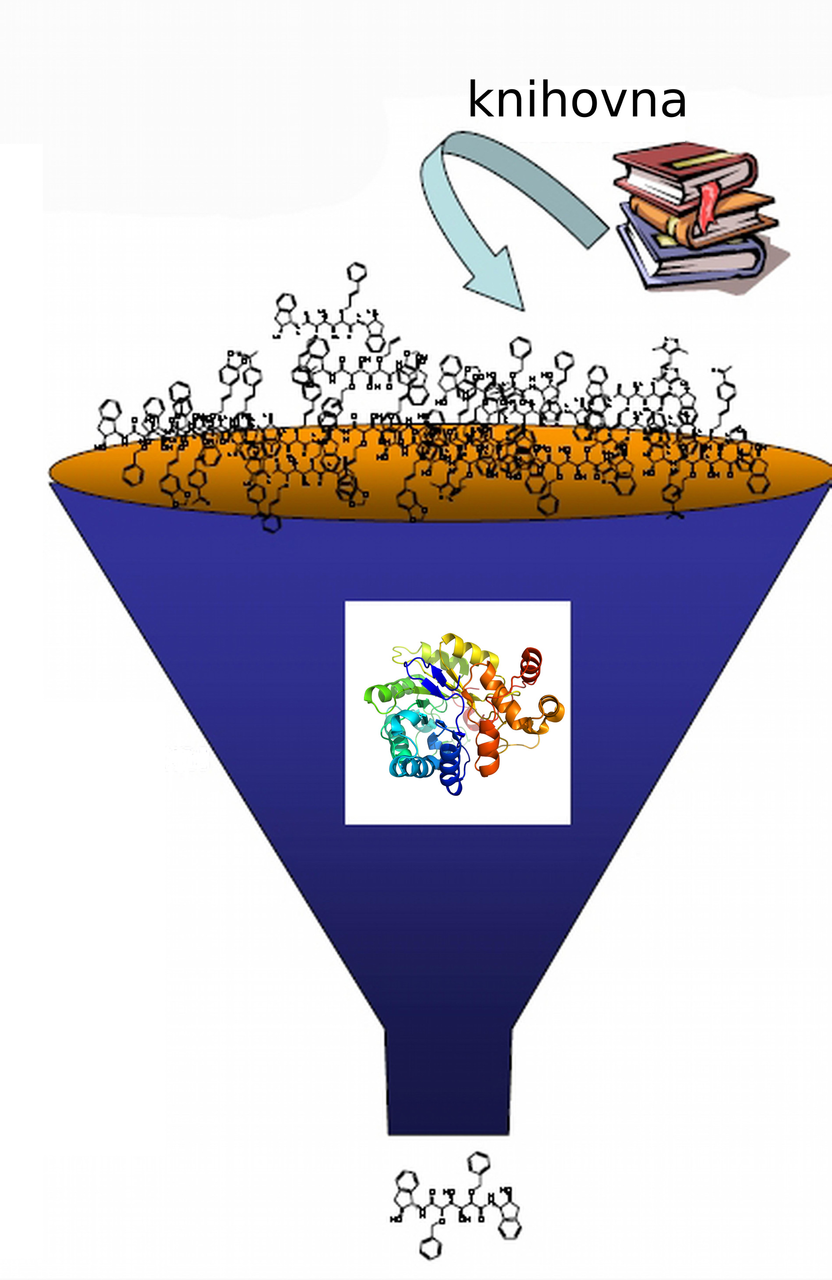
The Principal Investigator of the project focused on development of in silico drug design methods is Pavel Hobza from the Czech Academy of Sciences. This is his ninth project involving the use of a supercomputer, which was awarded IT4Innovations computational resources. The objective of his research group’s work is to develop a reliable computing strategy for identification of new ligands, which bind to therapeutically relevant proteins such as HIV protease, cyclin-dependent kinases, and aldo-keto reductases. The team is currently focused on developing reliable protocols for the virtual screening of compound libraries, which may contain millions of chemical substances. For the project of virtual screening for drug discovery, the team of professor Hobza was awarded more than 6 million core hours this time.
EFFECTS OF BIOMECHANICAL PROPERTIES OF LIPID MEMBRANES
Call: 14th Open Access Grant Competition
Researcher: Prof. Pavel Jungwirth
Institution: The Czech Academy of Sciences
Field: Material Sciences
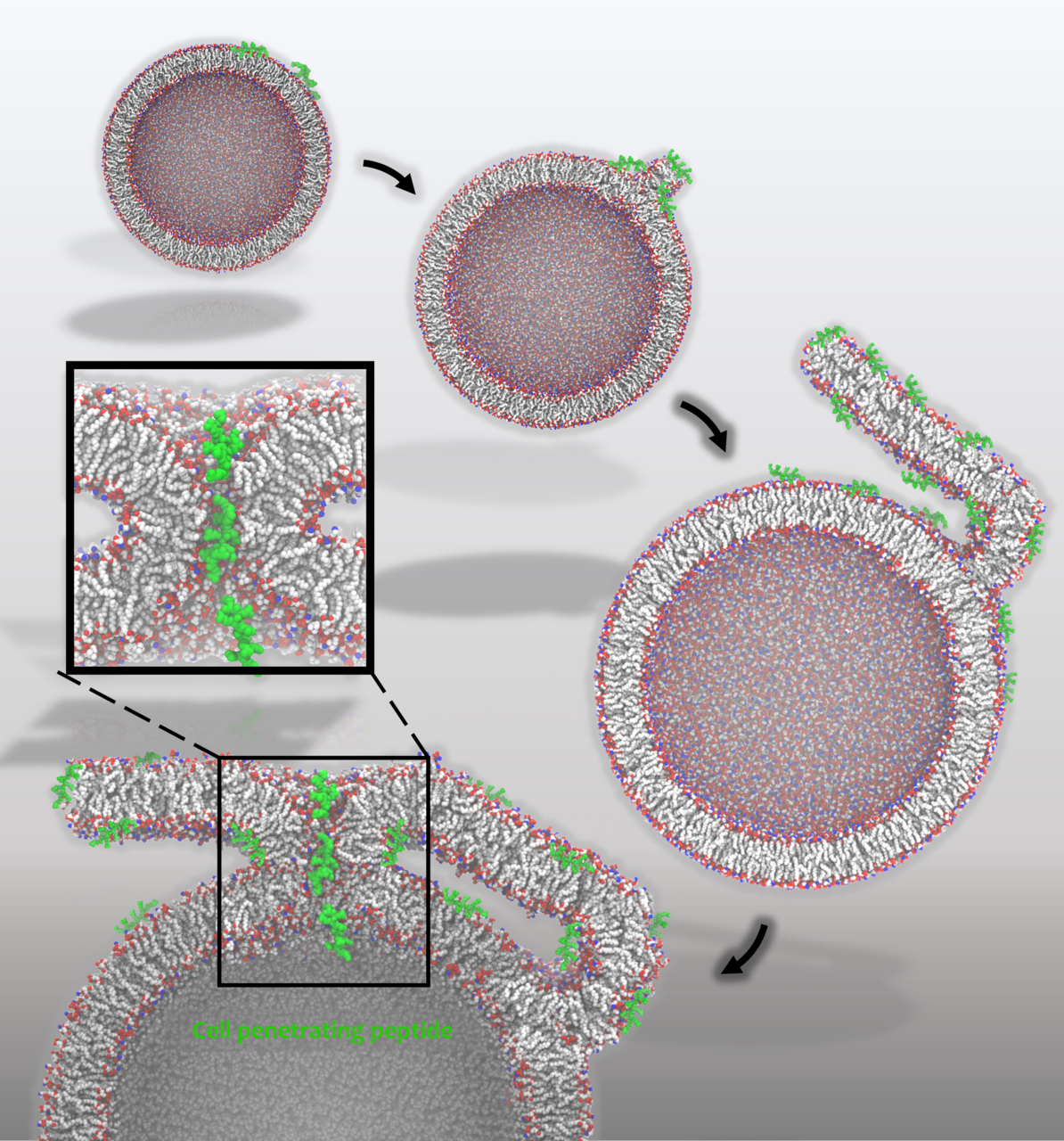
Prof. Pavel Jungwirth from the Institute of Organic Chemistry and Biochemistry of the CAS focuses on the research of macroscopic properties of lipid membranes. Aided by a supercomputer and molecular dynamics methods, he will perform a simulation of lipid bilayer behaviour. In plasma membranes, not only their chemical composition (e.g., types of the membrane-forming lipids) but also their shape will be investigated. The team of Prof. Jungwirth aims to detect the effects of the bilayer shape on membrane interactions. New findings about membrane shapes will open new opportunities for understanding the regulation of enzymes and other proteins in cells.
A MACHINE LEARNING APPROACH FOR THE DESCRIPTION OF ZEOLITES
Call: 14th Open Access Grant Competition
Researcher: Dr Miroslav Rubeš
Institution: Czech Academy of Sciences
Field: Material Sciences

The scientific domain with projects with the highest allocation of computational resources not only in our centre is material science. The project of Miroslav Rubeš from the Institute of Organic Chemistry and Biochemistry of the Czech Academy of Sciences, which was awarded almost 2 million core hours, falls in this domain as well. It is focused on zeolites, which are used as detergents, catalysts, and adsorbents. In 2017, the value of the global zeolite market was about 30 billion US dollars. The objective of Miroslav Rubeš project is to use machine learning algorithms to create a model which enables deeper understanding of the phenomena occurring in zeolitic materials.
ACCURACY AND PRECISION FOR MOLECULAR SOLIDS - II
Call: 13th Open Access Grant Competition
Researcher: Dr Jiří Klimeš
Institution: Charles University
Field: Material Sciences

Dr Jiří Klimeš and his research team were awarded almost 2 million core hours for their development of materials simulation methods. His “Accuracy and precision for molecular solids – II project” applies quantum chemistry knowledge and approaches used for the description of solids, and received the prestigious start-up grant by European Research Council. In nature as well as industry molecular solids (molecular crystals) play an important role, for example, methane hydrate, also called Burning Ice, a potentially very important source of energy, carbon dioxide ice caps on Mars, and pharmaceuticals in pills. Some molecular crystals have peculiar yet important properties. An interesting example is polymorphism, which is the ability of a molecule of the same compound to exist in different structures under same conditions, and as such it may be crucial for effectiveness of drugs in the body. The objective of Dr Klimeš‘ project is to develop methods for reliable calculations of lattice energies in materials such as molecular crystals, which will help understand their properties. In this project, the Salomon supercomputer will be used for extracting the lattice energies of 13 selected molecular crystals.
OPTIMIZATION DESIGN OF FUNCTIONAL MATERIALS IN A NEW TYPE OF LITHIUM BASED BATTERY
Call: 12th Open Access Grant Competition
Researcher: Dr Dominik Legut
Institution: IT4Innovations
Field: Material Sciences

Our colleague Dr. Dominik Legut is involved in the research of lithium-based metal batteries. Unlike lithium-ion batteries, these batteries have higher energy density and are capable of storing ten times more energy. However, lithium anodes face many of the challenges associated with their lower charging efficiency, change of volume while charging/ discharging, and especially with dendritic growth. In 2017, Dr. Legut, together with his colleagues from the USA, China, and Singapore, published an article about protective films for lithium-based metal batteries in the Advanced Energy Materials journal with an impact factor of 16. Special protective two-dimensional films with a thickness of a few atoms are capable of preventing electrodes connecting (and the subsequent dangerous short-circuit), which can potentially occur as a result of dendritic growth on lithium-based anodes. This time, Dr. Legut was awarded 8 million core hours for his research of the optimal structure of lithium-based anodes. Together with other colleagues, he will aim at designing optimal materials for lithium-based anodes using prediction algorithms, chemical stability and mechanical properties calculations.
DEVELOPMENT OF RELATIVISTIC SPECTROSCOPY (RESPECT) COMPUTATIONAL CODE FOR STUDY OF HEAVY METAL ANTICANCER COMPLEXES
Call: 12th Open Access Grant Competition
Researcher: Dr Jan Vícha
Institution: Tomáš Baťa University in Zlín
Field: Material Sciences

One of the cancer medical treatment methods is chemotherapy. The most frequently used chemotherapeutics are platinum-based drugs. The key step for their further development is a more detailed study of their structure, properties, dynamics, and reaction mechanisms. The project of Dr. Jan Vícha from Tomáš Baťa University in Zlín carries on his previous research project, as well as the results of his project, which was awarded computational resources within our 9th Open Access Grant Competition. The objective of the new project, which has now been awarded 1,134,000 core hours, is to enhance prediction ability and the accuracy of computation of complex platinum-based alloys spectroscopic properties using the ReSpect program developed by the project partner organization - Arctic University of Norway. The newly modified code of the program will first be tested using magnetic resonance parameter computations of simple platinum chemotherapeutics, such as cisplatin and oxaliplatin in solution. The scope of the research activities will then be extended to simulations of new advanced carriers of platinum drugs, which is also the main topic of the Advanced Carriers for Platinum Drugs project supported by the Grant Agency of the Czech Republic, also implemented by Dr. Vícha. The allocated computational resources will be used in testing the modified code and for relativistic quantum chemical computations performed by the ReSpect program for prediction and analysis of magnetic resonance parameters for heavy metal complex compounds.
MOLECULAR SIMULATIONS OF TIN BASED MATERIALS FOR EUV LITHOGRAPHY
Call: 11th Open Access Grant Competition
Researcher: Prof. Petr Slavíček
Institution: The University of Chemistry and Technology Prague
Field: Material Sciences

How can focused high-energy radiation change a material? What particular changes will occur at a molecular level? The chemical changes of materials under the influence of high- -energy photons are studied by the team led by Prof. Petr Slavíček from the Laboratory of the Theoretical Photodynamics Research Group at the University of Chemistry and Technology in Prague. Their project titled Molecular simulations of tin based materials for EUV lithography has been awarded 1,082,000 core hours. The objective of this project is to describe molecular changes occurring in extreme ultraviolet (EUV) ionization of tin based organic compounds (particularly so-called Sn-O cages). These compounds may potentially be used as photoresist materials for EUV lithography, a new generation of lithography for nanometric dimensions applicable in effective production of new computer chips. The method is based on the changes of physical and chemical properties of photoresists (e.g. their solubility) after EUV radiation. By exposing specific areas of a material to the radiation, the dimension of the resulting structure can be up to 10 nm, which is the threshold limit dimension of today‘s commercial chips. Considerable computational intensity of molecular simulations of ionized Sn-O compounds is generated by the rich electron structure of tin. Simulation of a single trajectory taking half a picosecond requires almost a week to be computed using common processors. With its 76,896 cores (Intel Haswell processors and Intel Xeon Phi accelerators), our Salomon supercomputer will allow the researchers to perform extensive simulation, which would not be practically executable otherwise.
IN SILICO DRUG DESIGN
Call: 10th Open Access Grant Competition
Researcher: Prof. Pavel Hobza
Institution: Institute of Organic Chemistry and Biochemistry of the Czech Academy of Sciences
Field: Material Sciences
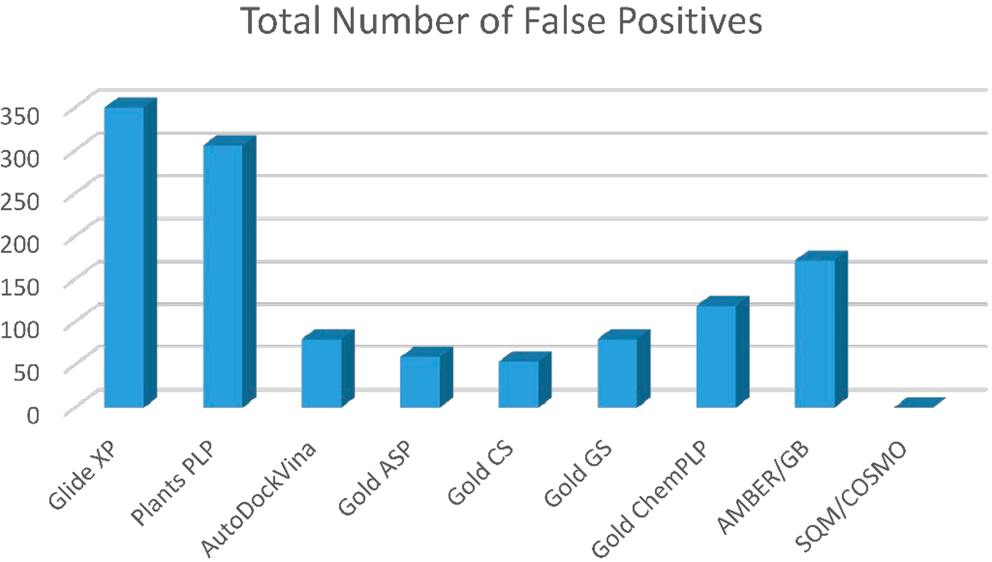
Prof. Pavel Hobza from the Institute of Organic Chemistry and Biochemistry of the Czech Academy of Sciences conducts research on computer-aided drug design. His project “In silico drug design” has been awarded 7,425,000 core hours within the 10th Open Access Competition. The allocated computational resources will be used for developing virtual screening methods for drugs. Used by the pharmaceutical industry, this approach is based on molecular modelling (docking and scoring) in order to identify suitable substances for designing new drugs. Due to its high computing performance requirements, the reliability of these methods has been low so far. Using the IT4Innovations supercomputers and employing exact quantum chemistry computations, the researchers from Prof. Hobza’s team are able to predict both the drug structure at the active site of proteins and their ability to bind, which determines their therapeutic effects. The recently published approach is currently used in collaboration with leading pharmaceutical companies.